Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124
Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124
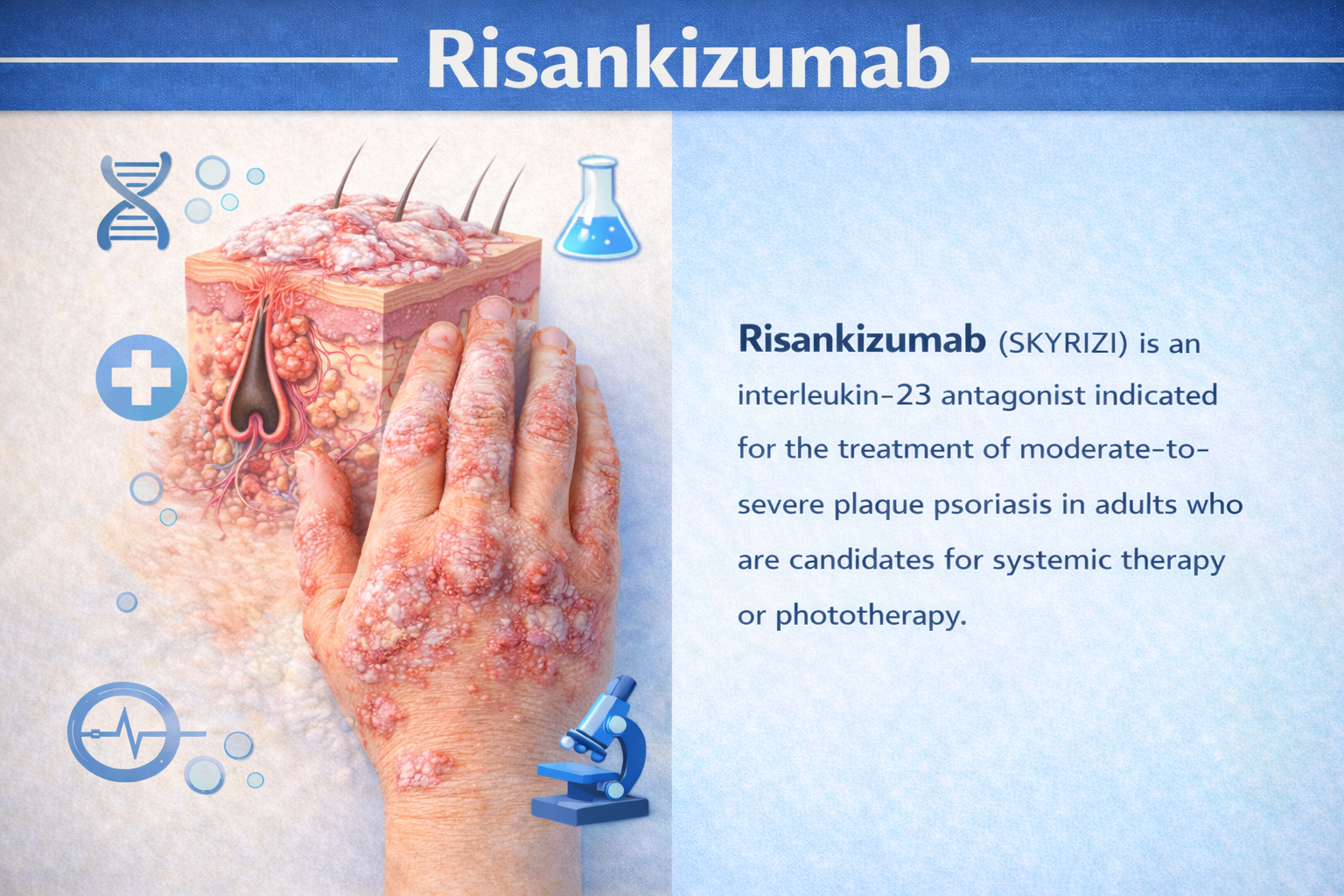
Clarity in Trials, Confidence in Decisions
Clarity in Trials, Confidence in Decisions
| Study | Design | Comparator | N (RZB) | Duration | Key Purpose |
|---|---|---|---|---|---|
| UltIMMa-1 (M16-008 / NCT02684370) | Ph3, RCT, DB, PBO + active-controlled | Placebo, ustekinumab | 304 | 52 weeks | Co-primary efficacy; maintenance |
| UltIMMa-2 (M15-995 / NCT02684357) | Ph3, RCT, DB, PBO + active-controlled | Placebo, ustekinumab | 294 | 52 weeks | Co-primary efficacy; maintenance |
| IMMhance (M15-992 / NCT02672852) | Ph3, RCT, DB; randomised withdrawal design | Placebo | 407 | 52+ weeks | Maintenance & durability; re-treatment |
| IMMvent (M16-010 / NCT02694523) | Ph3, RCT, DB; adalimumab switcher design | Adalimumab | 301 | 44 weeks | Head-to-head vs. TNFi; rescue switching |
| Phase 2 dose-ranging (1311.2) | Ph2, RCT, DB, dose-ranging | Ustekinumab | — | 24+ weeks | Dose selection (18 / 90 / 180 mg) |
RZB = risankizumab 150 mg SC; DB = double-blind; RCT = randomised controlled trial. Ustekinumab used in trials was EU-sourced; FDA noted the applicant did not provide a scientific bridge to US-licensed Stelara. Source: FDA Medical Review BLA 761105 (Ref ID: 4422774).
| Characteristic | UltIMMa-2 · RZB (N=294) | UltIMMa-2 · PBO (N=98) | UltIMMa-2 · UST (N=99) | UltIMMa-1 · RZB (N=304) | UltIMMa-1 · PBO (N=102) | UltIMMa-1 · UST (N=100) |
|---|---|---|---|---|---|---|
| Male | 203 (69%) | 67 (68%) | 66 (67%) | 212 (70%) | 79 (77%) | 70 (70%) |
| Mean Age (years) | 46.2 | 46.3 | 48.6 | 48.3 | 49.3 | 46.5 |
| Age ≥65 years | 28 (10%) | 12 (12%) | 17 (17%) | 36 (12%) | 16 (16%) | 12 (12%) |
| White | 255 (87%) | 87 (89%) | 91 (92%) | 200 (66%) | 71 (70%) | 74 (74%) |
| Asian | 25 (9%) | 7 (7%) | 4 (4%) | 86 (29%) | 28 (27%) | 22 (22%) |
| Body weight ≤100 kg | 203 (69%) | 67 (68%) | 69 (70%) | 226 (74%) | 76 (75%) | 74 (74%) |
| Prior TNF use | 67 (23%) | 26 (27%) | 24 (24%) | 67 (22%) | 22 (22%) | 19 (19%) |
UltIMMa-1 enrolled more Asian subjects than UltIMMa-2 owing to multi-country site distribution. Source: FDA Medical Review BLA 761105 (Ref ID: 4422774), Table 9.
| Parameter | UltIMMa-2 · RZB | UltIMMa-2 · PBO | UltIMMa-1 · RZB | UltIMMa-1 · PBO |
|---|---|---|---|---|
| sPGA Moderate (3) | 228 (78%) | 77 (79%) | 256 (84%) | 86 (84%) |
| sPGA Severe (4) | 66 (22%) | 21 (21%) | 48 (16%) | 16 (16%) |
| Mean PASI (SD) | 20.54 (7.83) | 18.86 (7.31) | 20.63 (7.68) | 20.50 (6.68) |
| Median PASI | 18.5 | 16.4 | 18.1 | 18.9 |
| Mean BSA% (SD) | 26.2 (15.9) | 23.9 (15.7) | 26.2 (15.4) | 27.9 (17.2) |
| Mean PSS (SD) | 8.27 (3.89) | 8.71 (3.46) | 7.99 (3.64) | 7.55 (3.37) |
PASI = Psoriasis Area and Severity Index; BSA = body surface area; PSS = Psoriasis Symptom Scale (0–16). Source: FDA Medical Review BLA 761105 (Ref ID: 4422774), Table 10.
| Parameter | IMMvent · RZB (N=301) | IMMvent · ADA (N=304) | IMMhance · RZB (N=407) | IMMhance · PBO (N=100) |
|---|---|---|---|---|
| sPGA Moderate (3) | 243 (81%) | 246 (81%) | 323 (79%) | 77 (77%) |
| sPGA Severe (4) | 58 (19%) | 58 (19%) | 84 (21%) | 23 (23%) |
| Mean PASI (SD) | 19.95 (7.46) | 19.72 (7.51) | 19.91 (7.94) | 21.17 (8.68) |
| Median BSA% | 21 | 20 | 19 | 23 |
| Prior TNF use | 44 (15%) | 45 (15%) | 150 (37%) | 35 (35%) |
IMMhance had a higher proportion of TNF-experienced subjects (~37%) compared to UltIMMa and IMMvent trials (~15–23%). Source: FDA Medical Review BLA 761105 (Ref ID: 4422774), Tables 19–20.
sPGA 0 or 1 (“clear or almost clear”)
PASI 90
All risankizumab vs. placebo p < 0.001. Source: FDA PI BLA 761105 (Ref ID: 4423209), Table 2; FDA Medical Review (Ref ID: 4422774), Table 11.
sPGA 0 or 1 (“clear or almost clear”)
PASI 90
All risankizumab vs. placebo p < 0.001. Source: FDA PI BLA 761105 (Ref ID: 4423209), Table 2; FDA Medical Review (Ref ID: 4422774), Table 12.
| Endpoint | UltIMMa-2 RZB (N=294) | UltIMMa-2 PBO (N=98) | UltIMMa-1 RZB (N=304) | UltIMMa-1 PBO (N=102) |
|---|---|---|---|---|
| sPGA 0 (“clear”) | 150 (51%) | 3 (3%) | 112 (37%) | 2 (2%) |
| PASI 100 | 149 (51%) | 2 (2%) | 109 (36%) | 0 (0%) |
| PSS 0 (“no symptoms”) | 92 (31%) | 0 (0%) | 89 (29%) | 2 (2%) |
| PASI 90 (vs. UST, N=99/100) | 220 (75%) | 47 (48%) | 229 (75%) | 42 (42%) |
| sPGA 0 or 1 (vs. UST, N=99/100) | 246 (84%) | 61 (62%) | 267 (88%) | 63 (63%) |
All comparisons vs. placebo p < 0.001. Risankizumab was superior to ustekinumab for all ranked secondary endpoints (p < 0.001). PSS = Psoriasis Symptom Scale. Source: FDA Medical Review BLA 761105 (Ref ID: 4422774), Table 12.
Risankizumab was statistically superior to adalimumab for both co-primary endpoints (p < 0.001). PASI 75 at Week 16: RZB 91% vs. ADA 72% (p < 0.001). PASI 100 at Week 16: RZB 40% vs. ADA 23% (p < 0.001). Source: FDA Medical Review BLA 761105 (Ref ID: 4422774), Tables 21–22.
Source: FDA PI BLA 761105 (Ref ID: 4423209), Section 14; FDA Medical Review (Ref ID: 4422774), Table 23.
| PRO Instrument | Finding | Timepoint |
|---|---|---|
| PSS 0 (“no symptoms”) | ~29–31% RZB vs. 0–2% PBO achieved PSS 0 in UltIMMa-1/2 (p < 0.001). PSS included in FDA labelling as COA-confirmed fit-for-purpose instrument. | Week 16 |
| DLQI 0 or 1 | ~66–67% RZB vs. ~4–8% PBO achieved DLQI 0/1 in UltIMMa-1/2. FDA declined to include DLQI in labelling due to content validity issues. | Week 16 |
| PSS domains (pain, redness, itching, burning) | Significant improvements in all PSS domain scores in risankizumab vs. placebo in both UltIMMa trials. | Week 16 |
Source: FDA PI BLA 761105 (Ref ID: 4423209), Section 14; FDA Medical Review BLA 761105 (Ref ID: 4422774).
| Adverse Drug Reaction | Risankizumab 150 mg (N=1306) n (%) | Placebo (N=300) n (%) | Notes |
|---|---|---|---|
| Upper respiratory infections | 170 (13.0%) | 29 (9.7%) | Viral/bacterial URI, sinusitis, rhinitis, nasopharyngitis, pharyngitis, tonsillitis |
| Headache | 46 (3.5%) | 6 (2.0%) | Includes tension, sinus, cervicogenic headache |
| Fatigue | 33 (2.5%) | 3 (1.0%) | Includes fatigue and asthenia |
| Injection site reactions | 19 (1.5%) | 3 (1.0%) | Bruising, erythema, pain, pruritus, swelling, warmth |
| Tinea infections | 14 (1.1%) | 1 (0.3%) | Tinea pedis, cruris, corporis, versicolor, manuum |
| Folliculitis | <1% (>0.1%) | Lower | Reported at <1% but >0.1%, at higher rate than placebo |
| Urticaria | <1% (>0.1%) | Lower | Reported at <1% but >0.1%, at higher rate than placebo |
Source: FDA PI BLA 761105 (Ref ID: 4423209), Table 1 (Section 6.1).
| Parameter | Risankizumab 150 mg | Comparator |
|---|---|---|
| SAEs at 16 weeks | 31/1306 (2.4%) | PBO: 12/300 (4.0%); UST: 12/199 (5.0%); ADA: 9/304 (3.0%) |
| SAEs at 52 weeks | 42/598 (7.0%) | UST: 18/199 (9.0%) |
| SAEs (full program) | 139/2234 (6.2%) [9.8 events/100 PY] | — |
| Discontinuations due to AE (16 wks) | 9 (0.7%) | PBO: 9 (3.0%) |
| Discontinuations due to AE (52 wks) | 5 (0.8%) | — |
| Deaths (initial submission) | 5 in risankizumab group; none attributed to drug | 2 in adalimumab group |
SAE = serious adverse event. Source: FDA Medical Review BLA 761105 (Ref ID: 4422774), Section 7.3.4.
| Population | Data & Recommendation |
|---|---|
| Pregnancy | Limited human data; insufficient to evaluate risk of major birth defects, miscarriage, or adverse maternal/fetal outcomes. IgG1 crosses placenta; fetal exposure possible. Animal data: dose-dependent fetal/infant loss in cynomolgus monkeys (32% at 5 mg/kg [2× MRHD] vs. 19% vehicle; 43% at 50 mg/kg [20× MRHD]). No teratogenicity or neurobehavioural effects. PMR issued for pregnancy registry. |
| Lactation | No data on presence of risankizumab-rzaa in human milk. Maternal IgG is present in human milk. Consider benefits of breastfeeding vs. risks. |
| Paediatric (<18 years) | Safety and efficacy not established. PMR issued for PK/safety/efficacy study in subjects 6 to <18 years (PREA). |
| Geriatric (≥65 years) | 243 subjects ≥65 years and 24 subjects ≥75 years in Phase 3 program. No overall differences in exposure, safety, or efficacy. Insufficient data to determine if elderly respond differently. |
| Renal / Hepatic impairment | No specific PK studies conducted. As a mAb, not expected to undergo significant renal elimination. No dose adjustment recommended. |
Source: FDA PI BLA 761105 (Ref ID: 4423209), Sections 8.1–8.5; FDA Medical Review (Ref ID: 4422774).
Source: FDA PI BLA 761105 (Ref ID: 4423209), Section 12.3.
| Parameter | Detail |
|---|---|
| Route / Form | Subcutaneous injection; two 75 mg/0.83 mL prefilled syringes per 150 mg dose |
| Bioavailability | 89% absolute bioavailability after SC injection |
| Tmax | 3–14 days post-SC injection |
| Distribution (Vss) | 11.2 L (CV 34%); consistent with restricted vascular/interstitial distribution typical of IgG1 mAbs |
| Parameter | Detail |
|---|---|
| Metabolism | Not formally characterised; expected catabolism to small peptides and amino acids via catabolic pathways similar to endogenous IgG |
| Clearance / t½ | CL 0.31 L/day (CV 24%); terminal t½ ~28 days |
| Steady state | Achieved by Week 16 following dosing at Weeks 0, 4, Q12W |
| Body weight effect | BW increases CL and Vd, decreasing plasma concentrations; response rates 3–7% lower in subjects >100 kg but no dose adjustment recommended |
| Population | PK Effect / Recommendation |
|---|---|
| Age (≥18 years) | No clinically significant differences in PK based on age. No dose adjustment. |
| Sex | No clinically significant PK differences reported. No dose adjustment. |
| Body weight | CL and Vd increase with body weight; plasma concentrations decrease. No dose adjustment recommended. |
| Renal impairment | No specific studies conducted. mAb catabolism not expected to undergo significant renal elimination. |
| Hepatic impairment | No specific studies conducted. |
| Paediatric (<18 years) | Not studied. PMR issued for PK study in ages 6 to <18 years. |
Source: FDA PI BLA 761105 (Ref ID: 4423209), Section 12.3.
| Study / Parameter | Findings |
|---|---|
| Binding affinity (human IL-23p19) | Kd <1 pM; no binding to human IL-12 at 1 µM; no binding to rat IL-23; Kd 15 nM for mouse IL-23 |
| STAT3 inhibition (DB cell line) | IC50 24 pM for IL-23-induced STAT3 phosphorylation; 13–36 pM range across assays |
| IL-17 inhibition (mouse splenocytes) | IC50 6.7 pM against human IL-23; does not inhibit IFN-γ induction by IL-12 (selectivity for p19 vs. p40) |
| Molecular features | Fc mutations (Leu234Ala, Leu235Ala) to minimise effector function; single N-linked glycosylation at Asn-297 (CH2 domain) |
Source: FDA Medical Review BLA 761105 (Ref ID: 4422774), Pharmacology/Toxicology section.
| Parameter | Recommendation |
|---|---|
| Injection technique | Subcutaneous; 45-degree angle insertion; gently pinch cleaned skin; push plunger rod fully; needle guard activates only when full dose delivered |
| Needle gauge / length | 29 gauge × ½ inch; fixed needle with needle guard in 1 mL glass syringe |
| Injection site rotation | Each of 2 injections per dose must be at a different site (≥1 inch apart). Rotate across thighs, abdomen, or (HCP only) upper outer arm across doses |
| Restricted sites | Do not inject into areas that are tender, bruised, erythematous, indurated, scarred, or affected by psoriasis |
| Storage after opening | Single-dose prefilled syringe — discard immediately after use; do not reuse. Dispose in FDA-cleared sharps container. |
| Formulation / storage | Refrigerate at 2°C–8°C (36°F–46°F); do not freeze; do not shake; keep in original carton to protect from light; not made with natural rubber latex |
| Population | Recommendation & Data |
|---|---|
| Pregnancy | Limited human data; insufficient to evaluate risk. IgG1 crosses placenta — fetal exposure possible. Animal data: dose-dependent fetal/infant loss (see §4.7). PMR issued for pregnancy registry. Not recommended unless clinical benefit outweighs risk. |
| Lactation | No data on presence in human milk. Maternal IgG1 is present in human milk. Consider benefits of breastfeeding vs. risk to infant. |
| Paediatric (<18 years) | Safety and efficacy not established. Do not use. |
| Geriatric (≥65 years) | No overall differences in exposure, safety, or efficacy vs. younger subjects. Use standard dosing. |
| Renal / Hepatic impairment | No specific studies. No dose adjustment recommended based on available data. |
Source: FDA PI BLA 761105 (Ref ID: 4423209), Sections 2.1, 2.2, 2.3, 4, 5.1–5.3, 8.1–8.5.
| Item | Detail |
|---|---|
| Application Type | BLA 351(a) — Biologics License Application (original) |
| BLA Number | 761105Orig1s000 |
| Applicant | AbbVie Inc., North Chicago, IL 60064 |
| Submission Date | April 23, 2018 |
| PDUFA Goal Date | April 23, 2019 |
| Actual Approval Date | April 23, 2019 |
| Review Division | ODE 3 / Division of Dermatology and Dental Products (DDDP) |
| Review Type | Standard Review |
| Breakthrough Therapy Designation | Not reported |
| Fast Track Designation | Not reported |
| Orphan Drug Designation | Not applicable (plaque psoriasis) |
| Advisory Committee | Not convened |
| REMS Required | No |
| Medical Review Ref ID | 4422774 |
| PI Ref ID | 4423209 |
| PMR / PMC | Study Type | Objective | Key Design Elements |
|---|---|---|---|
| PMR 1 — Pregnancy Registry (Prospective) | Registry-based observational cohort | Capture maternal, fetal, and infant outcomes in pregnant women exposed to risankizumab | Major/minor congenital malformations, spontaneous abortions, stillbirths, neonatal infections; follow through ≥1 year of infant life |
| PMR 2 — Pregnancy Registry (Retrospective) | Retrospective cohort or case-control (claims/EMR) | Assess congenital malformations, SAB, stillbirths in risankizumab-exposed pregnancies | Administrative claims or electronic medical record data; comparison to unexposed or disease-matched controls |
| PMR 3 — Long-term Safety / Malignancy | Observational cohort study | Long-term safety vs. other psoriasis therapies; primary outcome: malignancy | Secondary outcomes: serious infections, TB, OI, CV events, autoimmune, neurologic, GI/haematologic AEs; 4-year enrolment; ≥8-year follow-up |
| PMR 4 — Paediatric PK/Safety/Efficacy (PREA) | Interventional PK/safety/efficacy study | Evaluate PK, safety, and efficacy in children 6 to <18 years with moderate-to-severe plaque psoriasis | Minimum 1 year of risankizumab exposure; dose selection based on PK modelling |
| Domain | Note |
|---|---|
| Benefit-risk conclusion | FDA concluded the benefit-risk profile of risankizumab is favourable for the approved indication. Robust and consistent efficacy data across 4 Phase 3 trials; acceptable safety profile with manageable infection risk. |
| Labelling — PSS included | PSS (Psoriasis Symptom Scale) included in Section 14 as FDA COA-confirmed fit-for-purpose instrument. DLQI excluded due to content validity issues. |
| Labelling — Ustekinumab comparator | No head-to-head claim vs. ustekinumab in indication text. Comparative data presented descriptively only due to absence of scientific bridge between EU and US ustekinumab. |
| Immunogenicity labelling | ADA incidence (24% by Wk 52; 57% neutralising) and impact of high titers on PK/response included in PI §6.2 per FDA biologic labelling standards. |
| Pharmacovigilance | No REMS required. Standard pharmacovigilance through routine adverse event reporting (MedWatch). Long-term observational safety study (PMR 3) mandated. |
| Source documents | FDA PI: BLA 761105 (Ref ID: 4423209, April 2019). FDA Multi-Discipline Review: BLA 761105 (Ref ID: 4422774, April 2019). |