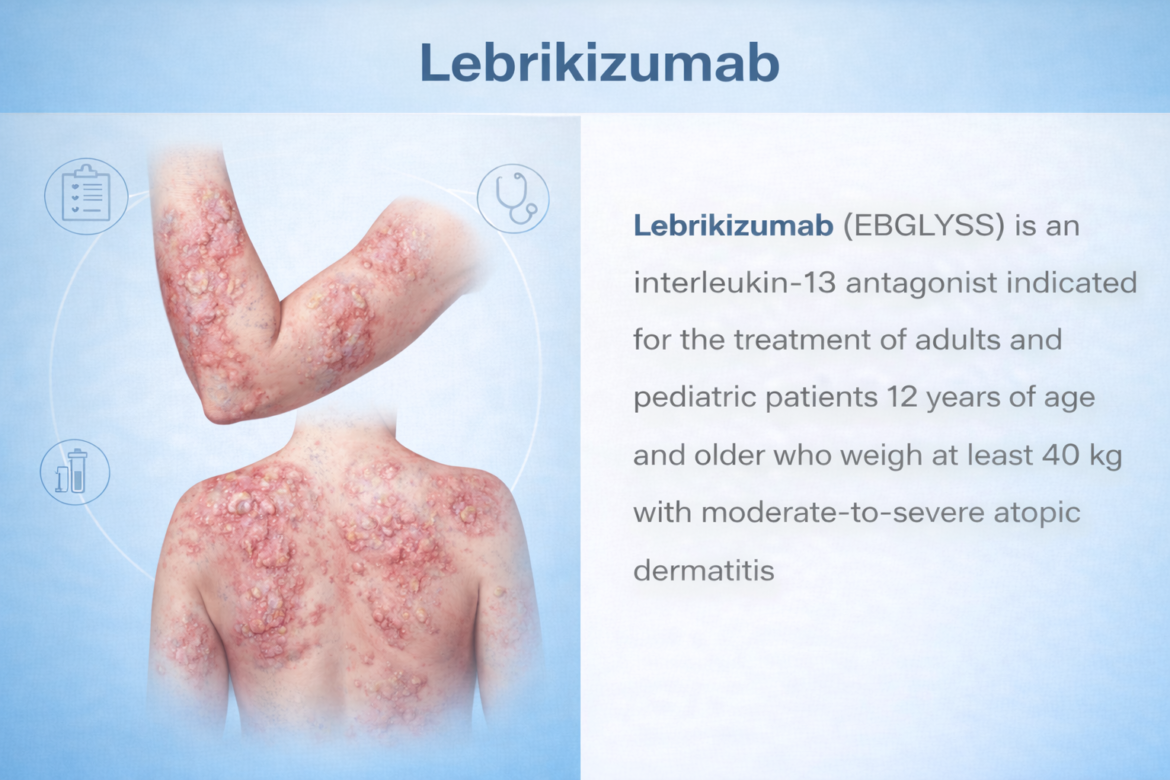
A fully human IgG4 monoclonal antibody that selectively binds IL-13 with high affinity and slow off-rate, blocking IL-4Rα/IL-13Rα1 signalling — a central driver of Type 2 skin inflammation in moderate-to-severe atopic dermatitis.
Application
BLA 761306
Target
Interleukin-13 (IL-13)
Route
Subcutaneous
Population
≥12 yrs · ≥40 kg
Half-life
~24.5 days
No Boxed Warning
✓ Clean safety profile
Drug Overview
01
IGA 0/1 at Week 16
43%
vs 13% placebo — ADvocate 1
EASI-75 at Week 16
59%
vs 16% placebo — ADvocate 1
Mechanism
Anti-IL-13 mAb
IgG4; blocks IL-4Rα/IL-13Rα1 signalling
Safety Database
1,756
subjects incl. 382 adolescents
Key Differentiator: Lebrikizumab binds IL-13 upstream, blocking the IL-4Rα/IL-13Rα1 heterodimeric complex while uniquely permitting IL-13/IL-13Rα2 engagement for natural IL-13 clearance. This distinguishes its receptor pharmacology from dupilumab (anti-IL-4Rα) and tralokinumab (anti-IL-13, different epitope).
Indication
EBGLYSS is indicated for the treatment of adults and pediatric patients 12 years of age and older who weigh at least 40 kg with moderate-to-severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. EBGLYSS can be used with or without topical corticosteroids.
Drug Identity
INN / Generic Name
Lebrikizumab-lbkz
Brand Name
EBGLYSS®
Manufacturer
Eli Lilly and Company, Indianapolis, IN 46285
Antibody Class
Fully human IgG4 monoclonal antibody
Target
Interleukin-13 (IL-13)
Molecular Weight
~145 kDa
Production
Chinese Hamster Ovary (CHO) cells via recombinant DNA technology
Formulation
Sterile, preservative-free; clear to opalescent, colorless to slightly yellow/brown; pH 5.4–6.0
Dosage Forms
250 mg/2 mL prefilled pen; 250 mg/2 mL prefilled syringe with needle shield
Storage
Refrigerate 2°C–8°C; may store ≤30°C up to 7 days; do not freeze
Pivotal Trial Program
Trial (Study ID)
NCT
Design
N
Duration
ADvocate 1 (KGAB)
NCT04146363
Phase 3, RCT, DB, PC; monotherapy
424
52 weeks
ADvocate 2 (KGAC)
NCT04178967
Phase 3, RCT, DB, PC; monotherapy
427
52 weeks
ADhere (KGAD)
NCT04250337
Phase 3, RCT, DB, PC; + TCS
211
16 weeks
Vaccine Response (KGAK)
—
Phase 3, RCT, DB, PC; vaccine substudy
254
16 weeks
DB = double-blind; PC = placebo-controlled; TCS = topical corticosteroids; Q2W = every 2 weeks. All Phase 3 trials enrolled subjects ≥12 years with moderate-to-severe AD (IGA ≥3, EASI ≥16, BSA ≥10%). Loading dose: 500 mg at Weeks 0 and 2.
Baseline Characteristics
02
Source: FDA Multi-Disciplinary Review, BLA 761306 (Reference ID 5252985). Demographic and disease characteristics reported from ITT (ADvocate 1) and mITT (ADvocate 2, ADhere) populations. All three trials enrolled subjects ≥12 years with IGA ≥3, EASI ≥16, and BSA ≥10%.
ADvocate 1 (KGAB)
424
LEB 250mg Q2W: N=283 · PBO: N=141
ADvocate 2 (KGAC)
427
LEB 250mg Q2W: N=281 · PBO: N=146
ADhere (KGAD)
211
LEB Q2W + TCS: N=145 · PBO + TCS: N=66
Total Enrolled
1,062
Across 3 Phase 3 pivotal trials
Age & Sex Distribution
Characteristic
ADvocate 1 (KGAB) · N=424
ADvocate 2 (KGAC) · N=427
ADhere (KGAD) · N=211
LEB Q2W (N=283)
PBO (N=141)
LEB Q2W (N=281)
PBO (N=146)
LEB+TCS (N=145)
PBO+TCS (N=66)
Mean Age, years (SD)
36.1 (17.8)
34.2 (16.4)
36.6 (16.8)
35.3 (17.2)
37.5 (19.9)
36.7 (17.9)
Median Age
31.0
30.0
32.0
30.0
33.0
32.0
Age Group: 12 to <18 yrs, n (%)
37 (13.1%)
18 (12.8%)
30 (10.7%)
17 (11.6%)
32 (22.1%)
14 (21.2%)
Age Group: 18 to <65 yrs, n (%)
225 (79.5%)
113 (80.1%)
228 (81.1%)
119 (81.5%)
98 (67.6%)
47 (71.2%)
Age Group: ≥65 yrs, n (%)
21 (7.4%)
10 (7.1%)
23 (8.2%)
10 (6.8%)
15 (10.3%)
5 (7.6%)
Female, n (%)
141 (49.8%)
73 (51.8%)
136 (48.4%)
75 (51.4%)
70 (48.3%)
33 (50.0%)
Male, n (%)
142 (50.2%)
68 (48.2%)
145 (51.6%)
71 (48.6%)
75 (51.7%)
33 (50.0%)
LEB = lebrikizumab; PBO = placebo; TCS = topical corticosteroids; SD = standard deviation. ADvocate 1 uses ITT population; ADvocate 2 and ADhere use mITT.
Race / Ethnicity — Pooled Trial Totals
ADvocate 1 (N=424)
White
68.2%
Asian
16.5%
Black / Af. Am.
11.6%
Am. Indian/AN
1.7%
Multiple / Other
~1.4%
ADvocate 2 (N=427)
White
59.3%
Asian
28.6%
Black / Af. Am.
8.2%
Am. Indian/AN
1.2%
Multiple / Other
~1.6%
ADhere (N=211)
White
61.6%
Asian
14.7%
Black / Af. Am.
13.3%
Am. Indian/AN
3.3%
Multiple / Other
~4.3%
Geographic Distribution — Top Enrolling Countries
Country
ADvocate 1 (N=424)
ADvocate 2 (N=427)
ADhere (N=211)
United States
190 (44.8%)
167 (39.1%)
151 (71.6%)
Poland
81 (19.1%)
—
27 (12.8%)
Germany
—
81 (19.0%)
11 (5.2%)
South Korea
34 (8.0%)
—
—
Australia
39 (9.2%)
—
—
Taiwan
—
60 (14.1%)
—
Canada
23 (5.4%)
58 (13.6%)
22 (10.4%)
Bulgaria
—
22 (5.2%)
—
Singapore
—
19 (4.4%)
—
Baseline Disease Characteristics
Baseline disease characteristics were well-balanced across treatment arms in all three trials. All subjects had moderate-to-severe AD (IGA ≥3, EASI ≥16, BSA ≥10%) at baseline, with inadequate response to topical therapies.
Disease Parameter
ADvocate 1 (KGAB)
ADvocate 2 (KGAC)
ADhere (KGAD)
LEB Q2W (N=283)
PBO (N=141)
LEB Q2W (N=281)
PBO (N=146)
LEB+TCS (N=145)
PBO+TCS (N=66)
IGA 3 (Moderate), n (%)
170 (60.1%)
83 (58.9%)
175 (62.3%)
95 (65.1%)
98 (67.6%)
48 (72.7%)
IGA 4 (Severe), n (%)
113 (39.9%)
58 (41.1%)
106 (37.7%)
51 (34.9%)
47 (32.4%)
18 (27.3%)
EASI — Mean (SD)
28.8 (11.3)
31.0 (12.9)
29.7 (12.0)
29.6 (10.8)
27.7 (11.1)
26.4 (10.6)
EASI — Median
25.7
26.7
26.0
27.6
25.1
23.7
EASI range (Min, Max)
16 – 72
16 – 67
16 – 70
16 – 65
16 – 72
16 – 66
Duration Since AD Onset
ADvocate 1 — Mean (SD)
22.6 yrs
15.0 SD · range 1–81 yrs; median 20.0
ADvocate 2 — Mean (SD)
20.5 yrs
14.8 SD · range 1–76 yrs; median 19.0
ADhere — Mean (SD)
21.1 yrs
16.3 SD · range 0–72 yrs; median 16.4
Adolescent Subgroup Summary
Adolescent enrollment (12–<18 years): 13.0% in ADvocate 1, 11.0% in ADvocate 2, and 21.8% in ADhere. Total of 372 pediatric subjects (12–<18 years) exposed across Phase 3 trials; 270 exposed ≥1 year. Safety and effectiveness were generally consistent with adult subjects. Stratification for randomisation included age group (adolescent vs adult) in both ADvocate trials.
Vaccine Response Study (KGAK) — Baseline Demographics
Study KGAK enrolled 254 adults (18–55 years) with moderate-to-severe AD in a 16-week Phase 3 randomized, double-blind, placebo-controlled study assessing vaccine immunogenicity. mITT population: N=247 (LEB: 125; PBO: 122). Two sites were excluded due to audit findings related to protocol non-compliance.
Characteristic
Lebrikizumab (N=125)
Placebo (N=122)
Age range
18–55 years
18–55 years
IGA ≥3 at baseline
Required
Required
EASI ≥16 at baseline
Required
Required
BSA ≥10% at baseline
Required
Required
Completed study, n (%)
113 (90.4%)
89 (73.0%)
Demographics balance
Comparable across arms per FDA review
Abbreviations: AD = atopic dermatitis; BSA = body surface area; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; ITT = intent-to-treat; LEB = lebrikizumab; mITT = modified intent-to-treat; PBO = placebo; Q2W = every 2 weeks; SD = standard deviation; TCS = topical corticosteroids. Source: FDA Clinical Review and Evaluation, BLA 761306 (Ref ID 5252985).
Clinical Efficacy
03
Efficacy evaluated at Week 16 across three Phase 3 trials. Primary endpoint: proportion achieving IGA 0 or 1 with ≥2-point improvement from baseline. Key secondaries: EASI-75, EASI-90, and ≥4-point improvement in Worst Daily Pruritus NRS. Subjects who received rescue therapy or discontinued were analysed as non-responders (MCMC-MI imputation).
Week 16 Primary Endpoint — IGA 0 or 1
ADvocate 1 (KGAB) — Monotherapy · N=424
500 mg loading at Wks 0 & 2, then 250 mg Q2W vs placebo — 16 weeks · ITT population
EBGLYSS 250 mg Q2W (N=283)
43%
Placebo (N=141)
13%
Δ +30.3% · p <0.0001
ADvocate 2 (KGAC) — Monotherapy · N=427
500 mg loading at Wks 0 & 2, then 250 mg Q2W vs placebo — 16 weeks · mITT population
All major secondary endpoints statistically significant vs placebo at p <0.0001, except Pruritus NRS at Week 2 in ADvocate 2 (p=0.02). Analysis: MCMC-MI imputation; CMH test adjusted for region, age group, and disease severity.
Efficacy Chart — Week 16 Response Rates
IGA 0/1 & EASI-75 Response Rates at Week 16
ADvocate 1 and ADvocate 2 — EBGLYSS 250 mg Q2W vs Placebo
Source: FDA Multi-Disciplinary Review BLA 761306 (Tables 28/29 and 31/32). MCMC-MI analysis.
Maintenance of Response — Week 52
Week 16 IGA/EASI-75 responders were re-randomised to 36 additional weeks of EBGLYSS 250 mg Q2W, 250 mg Q4W, or placebo.
Endpoint at Wk 52
ADv1: Q2W
ADv1: Q4W
ADv1: Placebo
ADv2: Q2W
ADv2: Q4W
ADv2: Placebo
IGA 0 or 1
76%
74%
47%
65%
81%
50%
EASI-75
79%
79%
61%
77%
85%
72%
N (IGA responders)
45
45
22
32
32
16
Safety & Adverse Reactions
04
Safety evaluated in 1,756 subjects across the Phase 3 programme including 382 adolescents; 891 treated for ≥1 year. Safety profiles were similar for monotherapy and concomitant TCS use.
Most Common Adverse Reactions — Induction Period (Wks 0–16)
Conjunctivitis (cluster)10% EBGLYSS vs 3% placebo (monotherapy). Mild–moderate; rarely led to discontinuation. Exposure-adjusted rate: 30.6/100 PY.
Injection Site Reactions3% EBGLYSS vs 1% placebo. Incidence declined with continued treatment. Includes pain, erythema, pruritus, swelling.
Herpes Zoster<1% EBGLYSS vs 0% placebo (3 subjects monotherapy; 2 with TCS). All non-serious.
Adverse Reactions Table (Weeks 0–16)
Adverse Reaction
EBGLYSS Q2W Mono (N=638)
Placebo Mono (N=338)
EBGLYSS Q2W+TCS (N=145)
Placebo+TCS (N=66)
Conjunctivitis (cluster)
10% (61)
3% (10)
5% (7)
0%
Injection Site Reactions (cluster)
3% (16)
1% (4)
3% (4)
2% (1)
Herpes Zoster
<1% (3)
0%
1% (2)
0%
Integrated analysis: ADvocate 1, ADvocate 2, and Phase 2 KGAF (monotherapy); ADhere (TCS concomitant). No boxed warning. No deaths in the placebo-controlled induction period attributed to lebrikizumab.
Warnings & Precautions
Hypersensitivity: Angioedema and urticaria reported. Discontinue EBGLYSS and initiate appropriate therapy if serious hypersensitivity occurs. Contraindicated in prior serious hypersensitivity to any component.
Conjunctivitis & Keratitis: Reported at higher rates vs placebo. Advise patients to report new-onset or worsening eye symptoms. Keratitis in 0.6% vs 0.3% placebo.
Parasitic (Helminth) Infections: Treat pre-existing infections before initiating. Discontinue if unresponsive to antihelminth treatment.
Live Vaccines: Avoid live vaccines immediately prior to or during treatment. Complete age-appropriate vaccinations before starting EBGLYSS.
Use in Special Populations
Population
Guidance
Pregnancy
No adequate data on risk. Monoclonal IgG crosses placenta; peak transfer in third trimester. No embryofetal toxicity in cynomolgus monkeys at 18× MRHD. Pregnancy registry PMR issued.
Lactation
No data on presence in human milk. Weigh benefits vs risks.
Pediatric (12–<18 yrs)
N=372 exposed; 270 ≥1 year. Safety and efficacy consistent with adults. Not established in patients <12 years or ≥12 years weighing <40 kg.
Lebrikizumab binds IL-13 upstream with high affinity and a slow off-rate, selectively blocking signalling through the IL-4Rα/IL-13Rα1 heterodimeric receptor complex — a central driver of Type 2 inflammation. Uniquely, IL-13 bound by lebrikizumab retains the ability to engage IL-13Rα2, facilitating natural IL-13 internalization and clearance.
PK Parameters
Bioavailability (SC)
~86%
Tmax ~7–8 days after single 250 mg SC dose
Volume of Dist. (Vss)
5.14 L
Limited extravascular distribution
Half-life (t½)
24.5 days
Linear elimination; dose-independent
Clearance
0.154 L/day
Linear; not dose-dependent (37.5–500 mg)
Steady State
Week 4
After loading doses at Wks 0 and 2
Metabolism
Catabolic
Degraded to peptides & amino acids; same as endogenous IgG
Steady-State Exposure by Regimen
Regimen
Cmax (mcg/mL)
Cavg (mcg/mL)
Ctrough (mcg/mL)
250 mg Q2W (induction / maintenance)
108
100
87
250 mg Q4W (maintenance)
63
51
36
PK in Special Populations
Factor
Effect on PK
Age
No significant effect
Sex
No significant effect
Race
No significant effect
Body Weight
Lower Ctrough in higher body weight; dose adjustment not required
Renal Impairment
No clinically significant differences (mild–moderate); not expected to undergo renal elimination
Hepatic Impairment
No studies conducted; not expected to undergo hepatic elimination as mAb
Dosing & Administration
06
Loading Dose
500 mg
2 × 250 mg SC at Weeks 0 & 2
Induction
250 mg Q2W
From Week 2 through response evaluation
Maintenance
250 mg Q4W
After adequate clinical response achieved
Route
SC
Subcutaneous injection
Dosing Schedule
Phase
Dose
Frequency
Duration
Loading
500 mg (2 × 250 mg injections)
Week 0 and Week 2
First 2 doses
Induction → Response Evaluation
250 mg
Every 2 weeks (Q2W)
Week 2 through Week 16 (or until adequate response)
Maintenance
250 mg
Every 4 weeks (Q4W)
After achieving IGA 0/1 or EASI-75 (Week 16 or later)
Adequate clinical response is defined as achieving IGA 0 or 1 or EASI-75. If not achieved at Week 16, continue Q2W until response is obtained. Transition to Q4W is individualised.
Contraindications
Prior serious hypersensitivity to lebrikizumab-lbkz or any excipients of EBGLYSS (glacial acetic acid, histidine, polysorbate 20, sucrose).
Regulatory History
07
Application
BLA 761306
Biologics License Application
Sponsor
Eli Lilly
Indianapolis, IN · US License No. 1891
US Approval
2024
Mod-to-severe AD, ≥12 yrs
Approval Timeline
Early Development
IL-13 Discovery & Early Clinical (Roche/Genentech)
Initially developed for asthma; proof of concept established in IL-13–driven disease.
Three multicenter RCTs enrolling 1,062 subjects ≥12 years with moderate-to-severe AD through Week 52.
Oct 2022
EMA Approval (Europe)
EBGLYSS approved in Europe for moderate-to-severe AD in adults.
Sep 2023
FDA Complete Response Letter
Deficiencies identified in drug substance/product manufacturing facilities. Not related to efficacy or safety.
Mar 2024
Resubmission (BLA 761306)
Applicant resubmitted addressing all CMC deficiencies; also submitted KGAK vaccine response study.
2024
FDA Approval — EBGLYSS® Granted
Approved for moderate-to-severe AD in adults and pediatric patients ≥12 years weighing ≥40 kg. Can be used with or without TCS.
May / Oct 2025
Labeling Updates
Warming instructions removed for prefilled pen (May 2025) and prefilled syringe (Oct 2025). Reference ID: 5684721.
Postmarketing Requirements (PMRs)
PMR
Requirement
Timeline
PMR 1
Prospective Pregnancy Exposure Registry — maternal, fetal & infant outcomes vs unexposed controls through age 1 year
Draft protocol: March 2025
PMR 2
Additional pregnancy study (different design — e.g. retrospective cohort or case-control) assessing major congenital malformations, SABs, stillbirths
Draft protocol: March 2025
PMR 3
RCT: PK and safety in pediatric patients 6 months to <18 years weighing <40 kg (3 age strata)
Ongoing at approval
PMR 4
Open-label long-term extension: safety in pediatric patients 6 months to <18 years weighing <40 kg
Ongoing at approval
Regulatory Context
Class context: Lebrikizumab is one of four biologics approved for moderate-to-severe AD as of 2024 (dupilumab, tralokinumab, lebrikizumab, nemolizumab). It is the only anti-IL-13 agent with a Q4W maintenance dosing option after initial Q2W induction, and the only IL-13 inhibitor permitting residual IL-13/IL-13Rα2 engagement for natural IL-13 clearance.