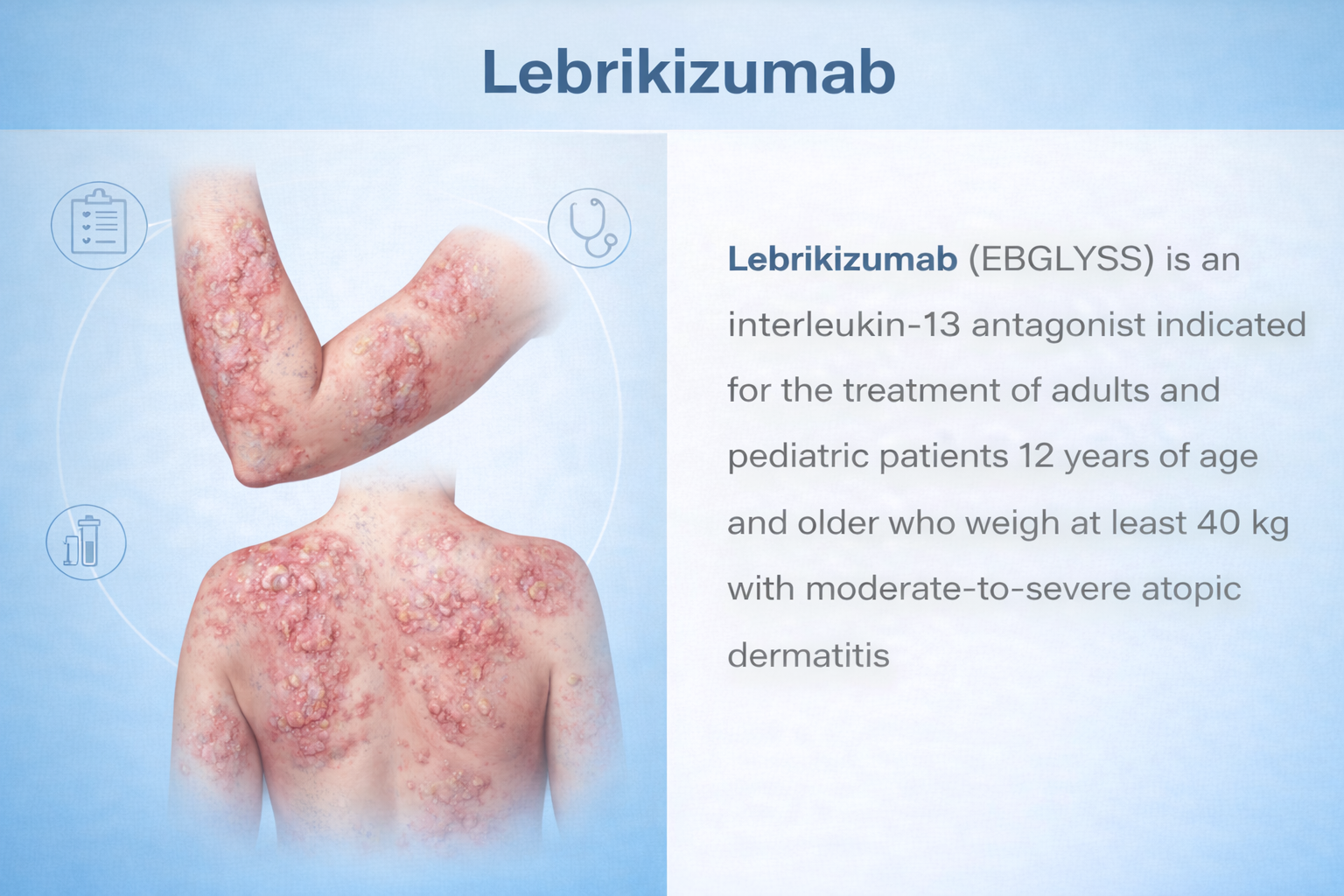
Indication
Moderate-to-Severe AD
Adults & pediatric ≥12 yrs, ≥40 kg; inadequate response to topical Rx
Mechanism
Anti-IL-13 IgG4 mAb
Blocks IL-4Rα/IL-13Rα1 signalling; permits IL-13Rα2 engagement
Dose & Schedule
500→250 mg SC
Loading Wks 0&2; Q2W induction; Q4W maintenance
Phase 3 Program
3 Trials
1,062 subjects · ADvocate 1, ADvocate 2, ADhere
Mechanism of Action: Lebrikizumab is an IgG4 monoclonal antibody that binds with high affinity and slow off-rate to interleukin-13 (IL-13), blocking signalling through the IL-4Rα/IL-13Rα1 heterodimeric receptor complex. IL-13 is a Type 2 cytokine central to atopic dermatitis pathogenesis; its inhibition reduces proinflammatory cytokines, chemokines, and IgE. Uniquely, lebrikizumab-bound IL-13 retains the ability to bind IL-13Rα2, enabling IL-13 internalization and natural clearance — a pharmacological distinction from anti-IL-4Rα agents such as dupilumab.
| Trial (Study ID) | NCT | Design | N Randomised | Duration | Comparator | Primary Endpoint |
|---|
| ADvocate 1 (KGAB) | NCT04146363 | Phase 3, R, DB, PC; monotherapy | 424 | 52 wks | Placebo SC | IGA 0/1 at Wk 16 |
| ADvocate 2 (KGAC) | NCT04178967 | Phase 3, R, DB, PC; monotherapy | 427 | 52 wks | Placebo SC | IGA 0/1 at Wk 16 |
| ADhere (KGAD) | NCT04250337 | Phase 3, R, DB, PC; + TCS | 211 | 16 wks | Placebo + TCS | IGA 0/1 at Wk 16 |
| KGAK (Vaccine study) | — | Phase 3, R, DB, PC; vaccine response | 254 | 16 wks | Placebo SC | Booster response to Tdap & MCV |
| KGAF (Phase 2 dose-ranging) | — | Phase 2, R, DB, PC | Not reported | Not reported | Placebo SC | IGA 0/1 at Wk 16 |
| Drug (INN + Brand) | Target | Formulation | US Approval | Approved Population |
|---|
| Lebrikizumab (EBGLYSS®) | IL-13 | SC prefilled pen/syringe | Sep 2024 | ≥12 yrs, ≥40 kg, mod-severe AD |
| Dupilumab (Dupixent®) | IL-4Rα (IL-4/IL-13) | SC prefilled syringe/pen | Mar 2017 | ≥6 months, mod-severe AD |
| Tralokinumab (Adbry®) | IL-13 | SC prefilled syringe | Dec 2021 | ≥12 yrs, mod-severe AD |
| Nemolizumab (Nemluvio®) | IL-31Rα | SC prefilled pen | Aug 2024 | ≥12 yrs, mod-severe AD |
| Upadacitinib (Rinvoq®) | JAK1 | Oral tablet | Jan 2022 | ≥12 yrs, mod-severe AD |
| Abrocitinib (Cibinqo®) | JAK1 | Oral tablet | Jan 2022 | ≥12 yrs, mod-severe AD |
| Proprietary Name | EBGLYSS® |
| INN / Generic Name | Lebrikizumab-lbkz |
| Code Name | LY3650150 |
| Manufacturer | Eli Lilly and Company, Indianapolis, IN 46285 · US License No. 1891 |
| Pharmacologic Class | Interleukin-13 (IL-13) antagonist; IgG4 monoclonal antibody |
| Molecular Weight | ~145 kDa |
| Production Cell Line | Chinese Hamster Ovary (CHO) cells; recombinant DNA technology |
| Dosage Forms & Strengths | 250 mg/2 mL single-dose prefilled pen; 250 mg/2 mL (125 mg/mL) single-dose prefilled syringe with needle shield |
| NDC — Prefilled Pen | 0002-7772-11 (carton of 1) |
| NDC — Prefilled Syringe | 0002-7797-11 (carton of 1) |
| Storage | Refrigerate 2°C–8°C; room temperature ≤30°C for up to 7 days; do not freeze; protect from light |
| Excipients | Glacial acetic acid (1.8 mg), histidine (6.2 mg), polysorbate 20 (0.6 mg), sucrose (119.6 mg), Water for Injection; pH 5.4–6.0 |
| Latex | Not made with natural rubber latex |
| Boxed Warning | None |
| PI Revision Date | October 2025 (Ref ID: 5684721) |
Trial Summary: Three pivotal Phase 3 trials enrolled a total of 1,062 subjects ≥12 years with moderate-to-severe AD (IGA ≥3, EASI ≥16, BSA ≥10%). ADvocate 1 used ITT population; ADvocate 2 and ADhere used mITT. All subjects had inadequate response to topical therapies. Baseline characteristics were generally well-balanced across arms. Source: FDA Multi-Disciplinary Review BLA 761306 (Ref ID 5252985), Tables 24–27.
ADvocate 1 (KGAB)
424
LEB Q2W: N=283 · PBO: N=141
ADvocate 2 (KGAC)
427
LEB Q2W: N=281 · PBO: N=146
ADhere (KGAD)
211
LEB+TCS: N=145 · PBO+TCS: N=66
Total Enrolled
1,062
Across 3 Phase 3 pivotal trials
Age & Sex Distribution
| Characteristic | ADv1 LEB Q2W (N=283) | ADv1 PBO (N=141) | ADv2 LEB Q2W (N=281) | ADv2 PBO (N=146) | ADhere LEB+TCS (N=145) | ADhere PBO+TCS (N=66) |
|---|
| Mean Age, years (SD) | 36.1 (17.8) | 34.2 (16.4) | 36.6 (16.8) | 35.3 (17.2) | 37.5 (19.9) | 36.7 (17.9) |
| Median Age, years | 31.0 | 30.0 | 32.0 | 30.0 | 33.0 | 32.0 |
| Age 12 to <18 yrs, n (%) | 37 (13.1%) | 18 (12.8%) | 30 (10.7%) | 17 (11.6%) | 32 (22.1%) | 14 (21.2%) |
| Age 18 to <65 yrs, n (%) | 225 (79.5%) | 113 (80.1%) | 228 (81.1%) | 119 (81.5%) | 98 (67.6%) | 47 (71.2%) |
| Age ≥65 yrs, n (%) | 21 (7.4%) | 10 (7.1%) | 23 (8.2%) | 10 (6.8%) | 15 (10.3%) | 5 (7.6%) |
| Female, n (%) | 141 (49.8%) | 73 (51.8%) | 136 (48.4%) | 75 (51.4%) | 70 (48.3%) | 33 (50.0%) |
| Male, n (%) | 142 (50.2%) | 68 (48.2%) | 145 (51.6%) | 71 (48.6%) | 75 (51.7%) | 33 (50.0%) |
Race — Pooled Distribution
Baseline Disease Characteristics
| Disease Parameter | ADv1 LEB (N=283) | ADv1 PBO (N=141) | ADv2 LEB (N=281) | ADv2 PBO (N=146) | ADhere LEB+TCS (N=145) | ADhere PBO+TCS (N=66) |
|---|
| IGA 3 (Moderate), n (%) | 170 (60.1%) | 83 (58.9%) | 175 (62.3%) | 95 (65.1%) | 98 (67.6%) | 48 (72.7%) |
| IGA 4 (Severe), n (%) | 113 (39.9%) | 58 (41.1%) | 106 (37.7%) | 51 (34.9%) | 47 (32.4%) | 18 (27.3%) |
| EASI — Mean (SD) | 28.8 (11.3) | 31.0 (12.9) | 29.7 (12.0) | 29.6 (10.8) | 27.7 (11.1) | 26.4 (10.6) |
| EASI — Median | 25.7 | 26.7 | 26.0 | 27.6 | 25.1 | 23.7 |
| Pruritus NRS — Mean (0–10) | 7.0 | 7.0 | 7.0 | 7.0 | Not reported by arm |
| AD Duration Mean (SD), yrs | 22.6 (15.0) | Not reported | 20.5 (14.8) | Not reported | 21.1 (16.3) | Not reported |
| Prior biologic use | 99% of all subjects had received prior treatment for AD |
| Asthma (comorbidity) | 30% across Phase 3 trials |
| Allergic rhinitis | 50% across Phase 3 trials |
| Food allergy | 31% across Phase 3 trials |
| Allergic conjunctivitis | 14% across Phase 3 trials |
Adolescent subgroup: 372 pediatric subjects (12–<18 years) were exposed across Phase 3 trials (13.0% in ADvocate 1, 11.0% in ADvocate 2, 21.8% in ADhere); 270 were exposed for ≥1 year. Safety and effectiveness were generally consistent with adult subjects. Randomisation was stratified by age group (adolescent vs adult).
Primary endpoint: Proportion of subjects achieving IGA 0 (clear) or 1 (almost clear) with ≥2-point improvement from baseline at Week 16. Statistical framework: Cochran-Mantel-Haenszel (CMH) test adjusted for region, age group, and disease severity. Missing data: MCMC Multiple Imputation (MCMC-MI). Subjects who received rescue therapy or discontinued for lack of efficacy were non-responders. Type 1 error controlled with hierarchical testing procedure. Significance threshold: p <0.05 (two-sided).
IGA 0 or 1 — Lebrikizumab 250 mg Q2W
Treatment difference: +30.3% (95% CI: 22%, 38%) · p <0.0001
EASI-75 — Lebrikizumab Q2W
EASI-90 — Lebrikizumab Q2W
Pruritus NRS ≥4-pt improvement — Lebrikizumab Q2W
Pruritus NRS ≥4-pt — Placebo
EASI-75 difference: +42% (95% CI: 33%, 51%); EASI-90: +29% (95% CI: 21%, 36%); Pruritus NRS ≥4pt: +33% (95% CI: 25%, 41%). All p <0.0001.
IGA 0 or 1 — Lebrikizumab 250 mg Q2W
Treatment difference: +22.4% (95% CI: 14%, 30%) · p <0.0001
EASI-75 — Lebrikizumab Q2W
EASI-90 — Lebrikizumab Q2W
Pruritus NRS ≥4-pt improvement — Lebrikizumab Q2W
Pruritus NRS ≥4-pt — Placebo
EASI-75 difference: +33% (95% CI: 24%, 42%); EASI-90: +21% (95% CI: 13%, 28%); Pruritus NRS ≥4pt: +28% (95% CI: 20%, 37%). All p <0.0001.
IGA 0/1 Response Rate Through Week 16
ADvocate 1 and ADvocate 2 — Lebrikizumab 250 mg Q2W vs Placebo
Source: FDA Multi-Disciplinary Review BLA 761306 (Ref ID 5252985), Figures 8 & 10; PI BLA 761306 (Ref ID 5684721), Figure 1. Data points at Weeks 0, 2, 4, 8, 16 per reported figures. Intermediate time points not explicitly reported set to null.
Subjects achieving IGA 0/1 or EASI-75 at Week 16 were re-randomised to EBGLYSS 250 mg Q2W, Q4W, or placebo for 36 additional weeks in ADvocate 1 and ADvocate 2.
IGA 0/1 at Wk 52 — ADv1: LEB Q2W (n=45)
IGA 0/1 at Wk 52 — ADv1: LEB Q4W (n=45)
IGA 0/1 at Wk 52 — ADv1: Placebo (n=22)
IGA 0/1 at Wk 52 — ADv2: LEB Q2W (n=32)
IGA 0/1 at Wk 52 — ADv2: LEB Q4W (n=32)
IGA 0/1 at Wk 52 — ADv2: Placebo (n=16)
Concomitant TCS Trial (ADhere) — Week 16
ADhere (KGAD) results were consistent with monotherapy trials. EBGLYSS + TCS vs placebo + TCS in subjects with moderate-to-severe AD. The efficacy and safety profile with concomitant TCS was consistent with the monotherapy trials. Specific response rates for ADhere are presented in the PI Table 3 (Ref ID 5684721) and FDA Medical Review Tables 33 (Ref ID 5252985).
Patient-Reported Outcomes
| PRO Instrument | Finding | Timepoint |
|---|
| Pruritus NRS ≥4-pt improvement | 45.9% LEB vs 13.0% PBO (ADv1); 39.8% vs 11.5% (ADv2) | Week 16 |
| Sleep-Loss NRS ≥4-pt improvement | 39.0% LEB vs 4.7% PBO (ADv1); 28.0% vs 8.2% (ADv2) | Week 16 |
| DLQI (Dermatology Life Quality Index) | Assessed as secondary endpoint; statistically superior to placebo | Week 16 |
| POEM (Patient-Oriented Eczema Measure) | Assessed as secondary endpoint; statistically superior to placebo | Week 16 |
No Boxed Warning. EBGLYSS (lebrikizumab) carries no boxed warning. The safety profile was characterised across a database of 1,756 subjects including 382 adolescents (12–<18 years); 891 subjects were treated for ≥1 year. Safety profiles were similar whether lebrikizumab was administered as monotherapy or with concomitant topical corticosteroids. Source: FDA Multi-Disciplinary Review BLA 761306 (Ref ID 5252985).
Safety Population (Controlled)
1,756
Subjects in Phase 3 programme; 382 adolescents
Long-Term Exposure (≥1 yr)
891
Subjects treated ≥1 year across development programme
Mean Age
37 yrs
50% male; 62% White; 20% Asian; 13% Black
Most Common ADR (≥1%)
Conjunctivitis
10% EBGLYSS vs 3% placebo in monotherapy
§4.1 Warnings & Precautions
- Hypersensitivity (§5.1): Angioedema and urticaria have been reported. Discontinue EBGLYSS and initiate appropriate therapy for serious hypersensitivity reactions. Prior serious hypersensitivity to lebrikizumab-lbkz or any excipients is a contraindication.
- Conjunctivitis and Keratitis (§5.2): Conjunctivitis occurred in 10% (EBGLYSS) vs 3% (placebo) in monotherapy trials at 16 weeks. Keratitis in 0.6% vs 0.3%. Most cases mild or moderate; recovered without treatment interruption. Advise patients to report new or worsening eye symptoms.
- Parasitic (Helminth) Infections (§5.3): IL-13 inhibition may alter immune response to helminths. Treat pre-existing infections before starting EBGLYSS. Discontinue if patients do not respond to antihelminth treatment during therapy.
- Vaccinations (§5.4): Avoid live vaccines immediately prior to or during treatment. Complete all age-appropriate vaccinations per current guidelines before initiation. Vaccine response study KGAK showed non-inferior humoral responses to Tdap and MCV.
§4.2 ADR Frequency Table — Weeks 0 to 16
| Adverse Reaction | EBGLYSS 250 mg Q2W Monotherapy (N=638) | Placebo Monotherapy (N=338) | EBGLYSS 250 mg Q2W + TCS (N=145) | Placebo + TCS (N=66) |
|---|
| Conjunctivitis (cluster)ᵃ | 61 (10%) | 10 (3%) | 7 (5%) | 0 |
| Injection Site Reactions (cluster)ᵇ | 16 (3%) | 4 (1%) | 4 (3%) | 1 (2%) |
| Herpes Zoster | 3 (<1%) | 0 | 2 (1%) | 0 |
§4.3 Serious Adverse Events (SAEs) — Induction Period
Deaths — Placebo-Controlled Induction Period
No deaths were reported among subjects receiving lebrikizumab during the placebo-controlled induction period. Four deaths were reported during extended exposure (1 during maintenance escape, 2 during long-term extension, 1 during open-label Study KGAE) — none considered likely related to study drug. Source: FDA Clinical Review (Ref ID 5446098).
SAEs — General Profile
SAE rates were low and comparable across arms in placebo-controlled periods. Malignancy-related events were infrequent; one breast cancer (Stage II) was reported in the KGAK vaccine study lebrikizumab arm, considered unrelated to study drug. Source: FDA Multi-Disciplinary Review (Ref ID 5252985), Tables 42–45.
Conjunctivitis as Serious Event
All 73 conjunctivitis events during the 16-week induction period were non-serious and mild or moderate in severity. Conjunctivitis led to treatment discontinuation in 3 subjects (induction) and 2 subjects (maintenance). Exposure-adjusted incidence rate: 30.6 events per 100 PY through Week 16.
Eosinophilia
Increased post-baseline blood eosinophils observed at higher frequency vs placebo. Eosinophilia (>5,000 cells/mcL) in 0.4% EBGLYSS vs 0% placebo through Week 16. Elevations were generally transient and did not result in discontinuation.
§4.4 Treatment Discontinuations Due to AEs
| Analysis Set | EBGLYSS (%) | Placebo (%) | Leading Reasons (EBGLYSS vs PBO) |
|---|
| Monotherapy trials (ADvocate 1, ADvocate 2, KGAF) — Wks 0–16 | 2.4% | 1.8% | Conjunctivitis/keratitis 0.6% vs 0.3%; injection site reactions 0.2% vs 0% |
| TCS trial (ADhere) — Wks 0–16 | 2.1% | 0% | Conjunctivitis 0.7% vs 0%; injection site reactions 0.7% vs 0% |
§4.5 Laboratory Abnormalities
| Laboratory Finding | EBGLYSS Q2W (N=638) | Placebo (N=338) | Notes |
|---|
| Eosinophilia (>5,000 cells/mcL) | 0.4% | 0% | Generally transient; no discontinuations |
| Hematology CTCAE Grade changes | See Medical Review Table 54 | — | No clinically significant neutropenia or thrombocytopenia identified |
| Hepatic safety TEAEs | See Medical Review Tables 76–79 | — | Low frequency; no signals identified |
| Blood pressure changes | See Medical Review Tables 55–56 | — | No clinically meaningful TE shifts identified |
§4.6 Immunogenicity
Anti-drug antibodies (ADA) to lebrikizumab-lbkz developed in 4/145 (2.8%) of subjects during the 12-month treatment period. Most of these antibodies were neutralising and of low titer. ADA development was not associated with changes to pharmacokinetics, efficacy, or safety. Similar ADA results were observed in pediatric subjects. Source: FDA PI BLA 761306 (Ref ID 5684721), §12.6.
§4.7 Special Populations Safety
| Population | Data & Guidance |
|---|
| Pregnancy | Insufficient data on drug-associated risk of birth defects or miscarriage. Monoclonal IgG actively transported across placenta — peaks in third trimester. No embryofetal toxicity or malformations in cynomolgus monkeys at plasma exposure ~18× MRHD. Pregnancy registry required (PMR 1–2). |
| Lactation | No data on presence in human milk, effects on breastfed infant, or milk production. Weigh benefits vs risks considering endogenous IgG transfer in human milk. |
| Pediatric (12–<18 yrs, ≥40 kg) | N=372 exposed; 270 ≥1 year. Safety and effectiveness consistent with adults. Not established in <12 years or ≥12 years weighing <40 kg (ongoing PMR studies). |
| Geriatric (≥65 yrs) | N=123 subjects ≥65 years; 29 subjects ≥75 years in clinical studies. Insufficient numbers to determine differential response vs younger adults. |
| Renal impairment | No clinically significant PK differences in mild or moderate renal impairment. Not expected to undergo significant renal elimination as a monoclonal antibody. No studies in severe renal impairment. |
| Hepatic impairment | No specific clinical pharmacology studies conducted. Not expected to undergo significant hepatic elimination. |
§4.8 Nonclinical Safety Summary
Carcinogenesis / Mutagenesis
Animal studies have not been conducted to evaluate carcinogenic or mutagenic potential. This is standard for biologics/monoclonal antibodies. Source: FDA PI §13.1.
Reproductive Toxicology — Female
No effects on reproductive organs, hormones, or menstrual cycle in sexually mature female cynomolgus monkeys at IV doses up to 25 mg/kg/week for 37 weeks (~15× MRHD by Cavg,ss).
Reproductive Toxicology — Male
No effects on reproductive organs or sperm analysis in sexually mature male cynomolgus monkeys at SC doses up to 25 mg/kg/week for 13 weeks (~11× MRHD by Cavg,ss).
Embryofetal / Prenatal Development
No malformations or embryofetal toxicity in cynomolgus monkeys at SC doses ~18× MRHD. No effects on morphological, functional, or immunological development in offspring from birth through 6 months.
§5.1 PK Parameters Grid
Bioavailability (%F, SC)
~86%
Absolute bioavailability following single 250 mg SC dose
Tmax (median)
7–8 days
After single SC 250 mg dose; injection site does not affect absorption
Volume of Dist. (Vss)
5.14 L
Steady-state volume; limited extravascular distribution
Half-life (t½)
24.5 days
Linear elimination; dose-independent over 37.5–500 mg
Clearance
0.154 L/day
Linear; independent of dose
Steady State
Week 4
Achieved after approved loading doses at Weeks 0 and 2
§5.2 Steady-State Exposure
| Regimen | Cmax (mcg/mL) | Cavg (mcg/mL) | Ctrough (mcg/mL) |
|---|
| 250 mg Q2W (after loading doses) | 108 | 100 | 87 |
| 250 mg Q4W (after loading doses) | 63 | 51 | 36 |
§5.3 Absorption
Following a single SC 250 mg dose, peak serum concentrations are achieved approximately 7–8 days post-dose. Absolute bioavailability is ~86%. Dose-proportional exposure has been demonstrated over the SC dose range of 37.5 to 500 mg. Injection site location (abdomen, thigh, upper arm) does not influence absorption. Source: PI §12.3.
§5.4 Distribution
The steady-state volume of distribution is 5.14 L, consistent with limited extravascular distribution typical of IgG monoclonal antibodies. As an IgG4 molecule, lebrikizumab is expected to distribute primarily to the systemic circulation and interstitial fluid.
§5.5 Metabolism & Elimination
| Parameter | Details |
|---|
| Metabolic pathway | Catabolism to small peptides and amino acids via proteolytic pathways, same as endogenous IgG. Not metabolised by cytochrome P450 enzymes. |
| Terminal half-life | 24.5 days |
| Clearance | 0.154 L/day; linear and dose-independent |
| Primary excretion | Not applicable for monoclonal antibodies (catabolism); not renally or hepatically eliminated |
| Neonatal Fc receptor (FcRn) | Standard IgG recycling expected via FcRn, as typical of IgG4 antibodies |
§5.6 Special Population PK
| Factor | Effect on PK | Dose Adjustment |
|---|
| Age | No significant effect | None required |
| Sex | No significant effect | None required |
| Race | No significant effect | None required |
| Body weight | Lower Ctrough in higher body weight subjects | None required; single dose for all weights ≥40 kg |
| Mild/moderate renal impairment | No clinically significant differences | None required |
| Severe renal impairment / ESRD | Not studied | Not expected to require adjustment (monoclonal antibody) |
| Hepatic impairment | Not studied | Not expected to require adjustment (monoclonal antibody) |
| Pediatric (12–<18 yrs) | Generally consistent with adults; weight-based effects apply | Same dose as adults (≥40 kg) |
§5.7 Drug–Drug Interactions
No formal DDI studies conducted. The effect of lebrikizumab on the pharmacokinetics of co-administered medications has not been studied. As a monoclonal antibody metabolised via catabolism rather than CYP450 pathways, clinically meaningful pharmacokinetic drug interactions are not expected. No dose adjustments for co-administered drugs are recommended based on mechanism. Source: FDA PI §12.3.
§5.8 Mechanism of Action
Lebrikizumab is an IgG4 monoclonal antibody that binds with high affinity and slow off-rate to interleukin-13 (IL-13). It allows IL-13 to bind to IL-13Rα1 but inhibits IL-13 signalling through the IL-4Rα/IL-13Rα1 heterodimeric receptor complex. IL-13 is a Type 2 cytokine involved in the inflammatory cascade central to atopic dermatitis pathogenesis. By blocking this signalling complex, lebrikizumab inhibits IL-13-induced proinflammatory cytokines, chemokines, and IgE. Notably, lebrikizumab-bound IL-13 retains the capacity to bind IL-13Rα2, enabling subsequent internalization and natural clearance of IL-13 — a mechanism distinct from anti-IL-4Rα agents. Source: FDA PI §12.1.
§5.9 Pharmacodynamics
In clinical studies, lebrikizumab reduced levels of the following Type 2 inflammation biomarkers:
| Biomarker | Full Name | Clinical Relevance |
|---|
| Serum periostin | Periostin (IL-13-induced matricellular protein) | Pharmacodynamic marker; not established as correlate of outcome |
| Total IgE | Immunoglobulin E | Reduced; clinical relevance not fully understood |
| CCL17 (TARC) | Thymus and activation-regulated chemokine | Type 2 inflammation marker; reduced with treatment |
| CCL18 (PARC) | Pulmonary and activation-regulated chemokine | Type 2 inflammation marker; reduced with treatment |
| CCL13 (MCP-4) | Monocyte chemotactic protein-4 | Type 2 inflammation marker; reduced with treatment |
Loading Dose
500 mg
Two 250 mg SC injections at Weeks 0 & 2
Induction
250 mg Q2W
From Week 2 until adequate response (Week 16+)
Maintenance
250 mg Q4W
After achieving IGA 0/1 or EASI-75
Route
SC
Subcutaneous injection only
Standard Dosing
- Loading: 500 mg (2 × 250 mg SC) at Week 0 and Week 2
- Induction: 250 mg SC every 2 weeks (Q2W) from Week 2 through Week 16 (or until adequate response)
- Maintenance: 250 mg SC every 4 weeks (Q4W) after achieving IGA 0/1 or EASI-75
- If adequate response not achieved at Week 16, continue Q2W until response obtained
Dose Modifications
- No dose adjustment for renal impairment (mild–moderate)
- No dose adjustment for hepatic impairment
- No age-based adjustment (adults and adolescents ≥12 yrs, ≥40 kg receive same dose)
- No weight-based adjustment within approved population (≥40 kg)
- Not approved for patients <12 years or patients ≥12 years weighing <40 kg
Preparation & Administration
- Administer subcutaneously — abdomen, thigh, or back of upper arm
- Alternate injection site with each injection
- Do not inject within 2 inches (5 cm) of navel or into affected skin
- Do not require warming to room temperature before use
- Inspect visually — clear to opalescent, colorless to slightly yellow/brown; do not use if cloudy or particulate
- Back of upper arm: caregiver or HCP only
- Adult patients may self-inject; caregivers may inject pediatric patients after training
Missed Dose
- Administer the dose as soon as possible after a missed dose
- Thereafter, resume dosing at the regular scheduled time
- Complete all age-appropriate vaccinations prior to initiating therapy
- Store refrigerated 2°C–8°C; room temperature ≤30°C for up to 7 days; do not freeze, shake, or microwave
Prior serious hypersensitivity to lebrikizumab-lbkz or any excipients of EBGLYSS (glacial acetic acid, histidine, polysorbate 20, sucrose, Water for Injection) is a contraindication. Source: FDA PI §4.
Vaccinations: Complete all age-appropriate vaccinations per current immunization guidelines prior to initiating EBGLYSS. Avoid live vaccines immediately prior to or during treatment. EBGLYSS may alter immunity and increase risk of infection following live vaccine administration.
| Parameter | Recommendation |
|---|
| Topical corticosteroids (TCS) | May be used with or without TCS. Efficacy established with and without TCS. |
| Topical calcineurin inhibitors (TCI) | May be used, but reserved for sensitive areas only: face, neck, intertriginous areas, and genital areas. |
| Concomitant immunosuppressants | Not studied in combination with other biologics or systemic immunosuppressants for AD. |
§7.1 BLA Key Facts
| Application Number | BLA 761306 / IND 119866 |
| Application Type | BLA 351(a) — Biologics License Application (new molecular entity) |
| Applicant | Eli Lilly and Company, Indianapolis, IN 46285 · US License No. 1891 |
| Original Submission Date | September 28, 2022 |
| Original PDUFA Goal Date | September 28, 2023 |
| Complete Response Letter | September 28, 2023 (CMC manufacturing deficiencies; no efficacy/safety issues) |
| Resubmission Date | March 14, 2024 |
| Resubmission PDUFA Date | September 14, 2024 |
| Actual Approval Date | September 2024 |
| Review Division / Office | Division of Dermatology and Dentistry / Office of Immunology and Inflammation (OII) |
| Review Type | Standard |
| Fast Track Designation | Yes — granted December 6, 2019 (requested October 8, 2019) |
| Breakthrough Therapy Designation | Not reported |
| Advisory Committee | Not required (no AdCom convened) |
| Medical Review Ref ID | 5252985 (original cycle); 5446098 (resubmission) |
| PI Ref ID | 5684721 |
§7.2 Regulatory Timeline
DECEMBER 10, 2014
IND 119866 Submitted
IND filed for the proposed indication of moderate-to-severe atopic dermatitis. IND-opening study was a Phase 2 RCT evaluating safety and efficacy in persistent moderate-to-severe AD inadequately controlled by topical corticosteroids.
JUNE 19, 2019
End-of-Phase 2 Meeting Held
FDA provided feedback on Phase 3 development programme including trial design, endpoints, safety database requirements, and adolescent enrollment.
JULY 18, 2019
Phase 3 Protocols Submitted (KGAB, KGAC)
Protocols for pivotal Phase 3 monotherapy studies ADvocate 1 and ADvocate 2 submitted to the Agency.
DECEMBER 6, 2019
Fast Track Designation Granted
Fast Track Designation granted for lebrikizumab in moderate-to-severe atopic dermatitis. Request submitted October 8, 2019.
DECEMBER 20, 2019
TCS Combination & Adolescent Study Protocols Submitted (KGAD, KGAE)
FDA advice letters issued December 11, 2019, and March 27, 2020, on secondary endpoints and missing data handling.
NOVEMBER 2019 — OCTOBER 2021
Pediatric Study Plan (iPSP) Agreed
Initial iPSP submitted November 11, 2019. Amended iPSP submitted March 24, 2021, covering patients 6 months to <18 years weighing <40 kg. Agency agreed October 21, 2021.
JUNE 13, 2022
Pre-BLA Meeting Held
Pre-BLA meeting covered Table of Contents, clinical data package, safety analyses, labelling claims, and maintenance dosing regimen.
SEPTEMBER 28, 2022
BLA 761306 Submitted
BLA submitted under 351(a) pathway for treatment of adults and adolescents ≥12 years with moderate-to-severe AD inadequately controlled by topical therapies. PDUFA goal date: September 28, 2023.
SEPTEMBER 28, 2023
Complete Response Letter (CRL) Issued
CRL issued due to deficiencies related exclusively to drug substance and drug product manufacturing facilities. Efficacy and safety evidence were established during original review cycle.
MARCH 14, 2024
BLA 761306 Resubmitted
Complete response addressing all CMC manufacturing deficiencies. Applicant also submitted KGAK vaccine response study data. Resubmission PDUFA date: September 14, 2024.
SEPTEMBER 2024
FDA Approval — EBGLYSS® Granted
Approved for moderate-to-severe atopic dermatitis in adults and pediatric patients ≥12 years weighing ≥40 kg, with or without topical corticosteroids. 4 PMRs issued.
MAY 2025 / OCTOBER 2025
Labelling Updates
Warming instructions removed for prefilled pen (May 2025) and prefilled syringe (October 2025). PI revised October 2025 (Ref ID: 5684721).
§7.3 Key Review Issues
CMC Manufacturing Deficiencies (CRL)
The original BLA received a Complete Response letter on September 28, 2023, due solely to deficiencies in drug substance and drug product manufacturing facilities. Efficacy and safety were considered established during the original review cycle. The Applicant resolved all CMC deficiencies in the March 2024 resubmission.
Pruritus NRS Statistical Analysis Plan
During the resubmission review, sensitivity analyses were conducted for pruritus NRS endpoints comparing the revised approach (≥4 out of 7 days for baseline, ≥1 out of 7 days for post-baseline) with the original SAP. Findings showed alignment between sensitivity and primary analyses with minimal impact on point estimates.
Vaccine Response Study (KGAK)
The Applicant submitted KGAK data showing non-inferior humoral responses to Tdap and MCV. However, the Agency determined that these results do not constitute clinical correlates of vaccine protection, and declined to incorporate KGAK results into prescribing information — consistent with evolving FDA policy on vaccine response studies for immunomodulatory products.
Pediatric Data & PREA Requirements
BLA submission triggered PREA requirements. The Applicant submitted protocols for two pediatric studies (6 months to <18 years, <40 kg). Both studies were ongoing at time of approval; 4 PMRs were issued covering pregnancy registries and pediatric populations.
§7.4 Postmarketing Requirements & Commitments
4 PMRs issued at approval. Two PMRs address pregnancy outcomes; two address pediatric use in age and weight ranges not covered by the approved indication.
| PMR | Study Type | Objective | Key Design Elements |
|---|
| PMR 1 | Prospective pregnancy exposure registry | Maternal, fetal, and infant outcomes in exposed vs unexposed women | Prospective observational cohort; detect major/minor congenital malformations, SABs, stillbirths, PTBs; infant follow-up ≥1 year. Draft protocol due: March 2025. |
| PMR 2 | Additional pregnancy study (different design) | Assess major congenital malformations, SABs, stillbirths, SGA, PTB | Retrospective cohort (claims/EMR) or case-control with outcome validation. Draft protocol due: March 2025. |
| PMR 3 | Randomised, DB, PC trial | PK and safety in pediatric patients 6 months to <18 years weighing <40 kg | Three age strata: 6 months to <6 years; 6 to <12 years; ≥12 to <18 years. Ongoing at approval. |
| PMR 4 | Open-label long-term extension study | Long-term safety in pediatric patients 6 months to <18 years weighing <40 kg | 6 months to <12 years and ≥12 to <18 years. Ongoing at approval. |
§7.5 Regulatory Notes
| Domain | Note |
|---|
| Benefit-risk conclusion | Benefit-risk profile unchanged since original review cycle. All CMC deficiencies resolved. Reviewer and Office Director recommended approval. Source: FDA Clinical Review (Ref ID 5446098). |
| REMS | No Risk Evaluation and Mitigation Strategy (REMS) required. |
| Immunogenicity labelling | ADA incidence 2.8% (4/145); most neutralising, low titer; no PK/efficacy/safety impact. Source: PI §12.6. |
| Statistical issues | Pruritus NRS sensitivity analysis (revised post-baseline data density requirement) conducted at FDA request; consistent with primary results. |
| Vaccine response labelling | KGAK results not incorporated into labelling per evolving FDA policy on vaccine response studies for immunomodulatory products. |
| Medical Review Ref IDs | Original cycle: 5252985; Resubmission: 5446098; Statistical Review: 5423049; PI: 5684721 |
Competitive / class context: Lebrikizumab is one of four biologics approved for moderate-to-severe AD in adults/adolescents as of September 2024 (alongside dupilumab, tralokinumab, and nemolizumab). It is a selective anti-IL-13 antibody — mechanistically distinct from dupilumab (anti-IL-4Rα) — and shares target class with tralokinumab but differs in epitope (lebrikizumab allows IL-13/IL-13Rα2 engagement and natural IL-13 clearance). It is the first anti-IL-13 agent approved with a Q4W maintenance option following Q2W induction, subject to demonstrating adequate response at Week 16.