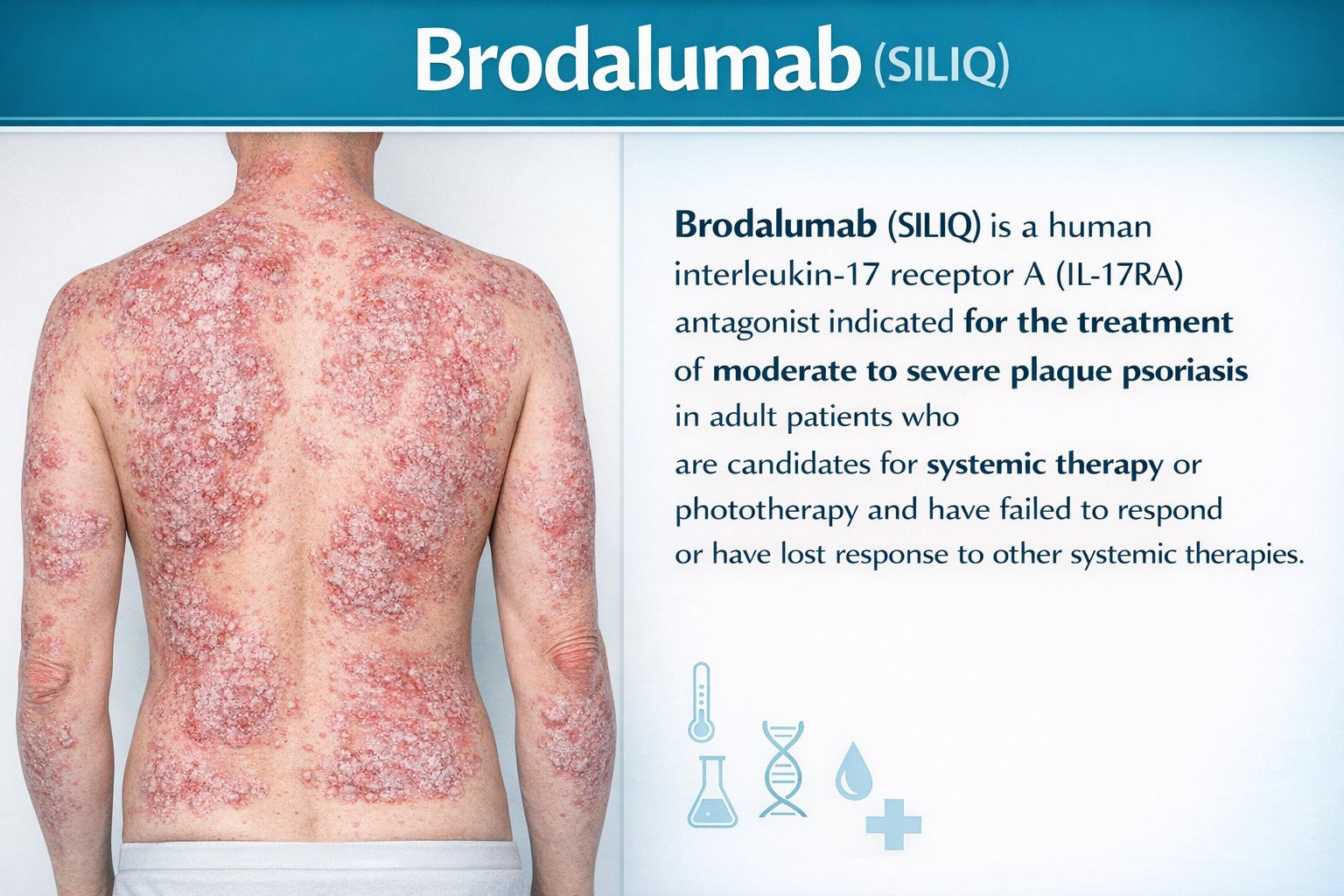
Indication
Moderate–Severe Plaque Psoriasis
Adults; candidates for systemic therapy/phototherapy; failed other systemic therapies
Mechanism
IL-17RA Antagonist
Human IgG2κ mAb — blocks IL-17A, IL-17F, IL-17C, IL-17A/F, IL-25
Dose & Schedule
210 mg SC
Wks 0, 1, 2 then every 2 weeks; reassess at 12–16 wks
Phase 3 Programme
3 Pivotal Trials
AMAGINE-1, -2, -3; total N = 4,373 subjects
⬛ BOXED WARNING — Suicidal Ideation and Behavior: Suicidal ideation and behavior, including 4 completed suicides, have occurred in patients treated with SILIQ. Prior to prescribing, weigh potential risks and benefits in patients with a history of depression and/or suicidal ideation or behavior. Patients with new or worsening suicidal thoughts and behavior should be referred to a mental health professional, as appropriate. SILIQ is available only through a restricted program under REMS called the SILIQ REMS Program.
Mechanism of Action: Brodalumab is a fully human monoclonal IgG2 antibody that selectively binds to human IL-17RA and inhibits its interactions with cytokines IL-17A, IL-17F, IL-17C, the IL-17A/F heterodimer, and IL-25. IL-17RA is a protein expressed on the cell surface and is a required component of receptor complexes utilised by multiple IL-17 family cytokines. Blocking IL-17RA inhibits IL-17 cytokine-induced responses including the release of pro-inflammatory cytokines and chemokines. Elevated levels of IL-17A, IL-17C, and IL-17F are found in psoriatic plaques; brodalumab’s broad receptor-level blockade distinguishes it from ligand-targeting approaches.
| Trial Name | NCT / Sponsor Code | Design | N Randomised (brodalumab 210 mg) | Duration | Comparator(s) | Primary Endpoint |
|---|
| AMAGINE-1 (Trial 1 / 20120104) | NCT01708590 | MC, R, DB, PC | 222 (vs 220 placebo) | 52 weeks (12-wk induction + RW phase) | Placebo | PASI 75 and sPGA 0/1 at Wk 12 |
| AMAGINE-2 (Trial 2 / 20120102) | NCT01708577 | MC, R, DB, PC, active-controlled | 612 (vs 300 uste; 309 pbo) | 52 weeks | Ustekinumab; Placebo | PASI 75, sPGA 0/1 at Wk 12 vs pbo; PASI 100 at Wk 12 vs ustekinumab |
| AMAGINE-3 (Trial 3 / 20120103) | NCT01708603 | MC, R, DB, PC, active-controlled | 624 (vs 313 uste; 315 pbo) | 52 weeks | Ustekinumab; Placebo | PASI 75, sPGA 0/1 at Wk 12 vs pbo; PASI 100 at Wk 12 vs ustekinumab |
| Drug (INN + Brand) | Target | Formulation | US Approval Date | Approved Population |
|---|
| Brodalumab (SILIQ) | IL-17RA (receptor) | 210 mg/1.5 mL SC prefilled syringe | February 2017 | Adults; failed other systemic therapies; REMS required |
| secukinumab (Cosentyx) | IL-17A (ligand) | 150 mg/1 mL SC prefilled syringe/pen | January 2015 | Adults — moderate-to-severe plaque psoriasis |
| ixekizumab (Taltz) | IL-17A (ligand) | 80 mg/1 mL SC autoinjector/syringe | March 2016 | Adults — moderate-to-severe plaque psoriasis |
| ustekinumab (Stelara) | IL-12/IL-23 (p40) | 45 mg or 90 mg SC injection | September 2009 | Adults — moderate-to-severe plaque psoriasis |
| Parameter | Details |
|---|
| Proprietary Name | SILIQ™ |
| INN / Generic Name | brodalumab |
| Code Names | AMG 827; KHK4827 |
| Manufacturer | Valeant Pharmaceuticals Luxembourg S.à.r.l., Grand Duchy of Luxembourg, L-1931 (manufactured for Valeant Pharmaceuticals North America LLC, Bridgewater, NJ 08807). U.S. License No. 2053. |
| Pharmacologic Class | Human interleukin-17 receptor A (IL-17RA) antagonist; human IgG2κ monoclonal antibody |
| Dosage Forms & Strengths | Injection: 210 mg/1.5 mL solution in a single-dose prefilled syringe |
| NDC | 0187-0004-02 (carton of two 210 mg/1.5 mL single-dose prefilled syringes) |
| Storage | Refrigerated 2°C to 8°C (36°F to 46°F) in original carton. If needed, may store at room temperature up to 25°C (77°F) for maximum 14 days; do not return to refrigerator once at room temperature. Do not freeze. Do not shake. |
| Inactive Ingredients | Glutamate (6.5 mg), polysorbate 20 (0.15 mg), proline (36 mg), and Water for Injection, USP at pH 4.8 |
| Molecular Formula & MW | 1312 amino acids; estimated molecular mass ~144,000 Daltons (IgG2κ mAb expressed in CHO cell line) |
| Boxed Warning | PRESENT — Suicidal Ideation and Behavior (see §4) |
| PI Revision Date | February 2017 (Ref ID: 4056898) |
Pooled Pivotal Trials (AMAGINE-1, -2, -3): Three multicenter, randomised, double-blind, controlled trials enrolled a total of 4,373 subjects. The overall safety population (controlled + open-label extension) comprised 4,558 subjects: brodalumab 210 mg n=3,066; ustekinumab n=613; placebo n=879. Baseline demographics were generally consistent across all treatment groups and all three trials. Source: FDA Medical Review BLA 761032 (Ref ID 4056898).
Total Pivotal Enrolment
4,373
Across AMAGINE-1, -2, -3 (pivotal trials)
Mean Age
45 years
Predominantly 40–64 years (58%)
Male Sex
69%
Consistent across all arms
Prior Biologic Use
~30%
~12% failed previous biologic therapy
| Parameter | Brodalumab 210 mg (N=3,066) | Ustekinumab (N=613) | Placebo (N=879) |
|---|
| Male sex | 69% | 68% | 69% |
| Mean age, years (SD) | 44.8 (13) | 45.1 (13) | 44.6 (13) |
| Age ≥65 years | 6% | 7% | 6% |
| White race | 91% | 90% | 91% |
| Mean body weight (SD), kg | 90.5 kg (mean reported) | Not reported separately | Not reported separately |
| Body weight >100 kg | 28% | Not reported | Not reported |
| Median baseline PASI score | 17.4 | 17.4 | 17.4 |
| Baseline PASI range | 9.4 – 72 | Not reported | Not reported |
| Median baseline BSA affected, % | 21% | Not reported | Not reported |
| BSA affected range, % | 10 – 97% | Not reported | Not reported |
| sPGA 3 (moderate) at baseline | 58% | Not reported | Not reported |
| sPGA 5 (very severe) at baseline | 5% | Not reported | Not reported |
| Psoriatic arthritis history | 21% | 19% | 21% |
| Prior biologic therapy | ~30% | ~26% | ~30% |
| Failed prior biologic therapy | 12% | Not reported | Not reported |
| Psychiatric disorder (any) at baseline | 18% | 20% | 17% |
| Depression at baseline | 14% | 16% | 13% |
Co-primary endpoints (all 3 trials vs placebo): (1) PASI 75 — proportion of subjects achieving ≥75% reduction in PASI composite score; (2) sPGA 0/1 (clear or almost clear) with ≥2-point improvement from baseline — both at Week 12. Additional primary endpoint (AMAGINE-2 and AMAGINE-3 vs ustekinumab): PASI 100 (complete clearance) at Week 12. Statistical framework: Non-Responder Imputation (NRI). All comparisons vs placebo: p<0.001. PASI 100 comparisons vs ustekinumab: p<0.001 in both AMAGINE-2 and AMAGINE-3. Familywise error rate (FWER) controlled by hierarchical testing procedure.
PASI 75 — brodalumab 210 mg (co-primary)
sPGA 0/1 — brodalumab 210 mg (co-primary)
PASI 100 — brodalumab 210 mg
sPGA 0 (clear) — brodalumab 210 mg
All comparisons vs placebo p<0.001. Source: PI Table 2, BLA 761032 (Ref ID 4056898).
PASI 75 — brodalumab 210 mg (co-primary)
sPGA 0/1 — brodalumab 210 mg (co-primary)
PASI 100 — brodalumab 210 mg (vs uste primary)
sPGA 0 (clear) — brodalumab 210 mg
sPGA 0 (clear) — Ustekinumab
PASI 100 vs ustekinumab: p<0.001. All comparisons vs placebo: p<0.001. Source: PI Table 2, BLA 761032 (Ref ID 4056898).
PASI 75 — brodalumab 210 mg (co-primary)
sPGA 0/1 — brodalumab 210 mg (co-primary)
PASI 100 — brodalumab 210 mg (vs uste primary)
sPGA 0 (clear) — brodalumab 210 mg
sPGA 0 (clear) — Ustekinumab
PASI 100 vs ustekinumab: p<0.001. All comparisons vs placebo: p<0.001. Source: PI Table 2, BLA 761032 (Ref ID 4056898).
| Trial / Endpoint | Brodalumab 210 mg Q2W (continued) | Placebo / Withdrawal |
|---|
| AMAGINE-1: sPGA 0/1 at Week 52 (among Wk 12 responders) | 83% (69/83) | 0% (0/84) |
| AMAGINE-1: PASI 75 at Week 52 (among Wk 12 responders) | 87% (72/83) | 0% (0/84) |
| AMAGINE-2 & -3: sPGA 0/1 at Week 52 (among Wk 12 responders) | 79% | Not applicable (re-randomised design) |
| AMAGINE-2 & -3: PASI 100 at Week 52 (among Wk 12 PASI 100 responders) | 72% | Not applicable |
| PRO Instrument | Finding | Timepoint |
|---|
| Psoriasis Symptom Inventory (PSI) — all 8 items at 0/1 score | Greater proportion of subjects in brodalumab 210 mg Q2W group achieved PSI score of 0 (not at all) or 1 (mild) on every item (itch, redness, scaling, burning, stinging, cracking, flaking, and pain) compared to placebo | Week 12 |
⬛ BOXED WARNING — Suicidal Ideation and Behavior: Suicidal ideation and behavior, including 4 completed suicides, occurred in subjects treated with SILIQ in the psoriasis clinical trials. There were no completed suicides in the 12-week placebo-controlled portion of the trials. SILIQ users with a history of suicidality or depression had an increased incidence of suicidal ideation and behavior as compared to users without such a history. A causal association between treatment with SILIQ and increased risk of suicidal ideation and behavior has not been established. Prior to prescribing SILIQ, weigh the potential risks and benefits in patients with a history of depression and/or suicidal ideation or behavior. SILIQ is available only through a restricted program under a REMS called the SILIQ REMS Program.
Safety Population (Controlled)
3,066
Brodalumab subjects in controlled trials (+ 879 pbo, 613 uste)
Long-term Exposure
3,755
Subjects exposed to brodalumab ≥1 year (total 4,464 received ≥1 dose)
Discontinuations (Wks 0–12)
~1%
All treatment groups; reasons: neutropenia, arthralgia, urticaria
Most Common ADR (≥1%)
Arthralgia
4.7% brodalumab vs 3.3% placebo at Week 12
Suicidal Ideation and Behavior (Boxed Warning): 34/4,464 subjects (0.37 per 100 subject-years) across all psoriasis clinical trials; 4 completed suicides. 8/10 who attempted or completed suicide had prior history of depression/suicidality. SILIQ REMS mandatory. Weigh benefit-risk in patients with depression history; refer to mental health professional if symptoms emerge.
Contraindication — Crohn’s Disease: Crohn’s disease occurred in one subject during psoriasis trials; exacerbation observed in other trials. Contraindicated in Crohn’s disease. Discontinue if Crohn’s develops during treatment.
Serious Infections: Subjects treated with brodalumab had higher rate of serious infections than placebo (0.5% vs 0.2%) and higher rates of fungal infections (2.4% vs 0.9%) overall. One case of cryptococcal meningitis during 12-week randomised period led to discontinuation. Consider risk-benefit in patients with chronic or recurrent infection. Evaluate for TB prior to initiation.
Tuberculosis (TB): Evaluate all patients for TB infection prior to initiating brodalumab. Do not administer to patients with active TB. Initiate treatment for latent TB prior to administering brodalumab. Consider anti-TB therapy in patients with past history of latent/active TB if adequate treatment cannot be confirmed.
Neutropenia: During 12-week randomised period, neutropenia occurred in 0.7% of subjects. Grade ≥3 (<1000/mm³): 0.5% brodalumab vs 0.2% ustekinumab vs 0% placebo. ANC reduction in 6.8% brodalumab vs 3.3% ustekinumab and 3.6% placebo. No serious infections associated with neutropenia cases.
Immunizations: Avoid use of live vaccines in patients treated with brodalumab. No data available on the ability of live or inactive vaccines to elicit an immune response during treatment.
| Adverse Reaction | Placebo (N=879) n (%) | Brodalumab 210 mg Q2W (N=1,496) n (%) | Ustekinumab (N=613)ᵇ n (%) |
|---|
| Arthralgia | 29 (3.3%) | 71 (4.7%) | 15 (2.4%) |
| Headache | 31 (3.5%) | 64 (4.3%) | 23 (3.8%) |
| Fatigue | 10 (1.1%) | 39 (2.6%) | 16 (2.6%) |
| Diarrhea | 10 (1.1%) | 33 (2.2%) | 5 (0.8%) |
| Oropharyngeal pain | 10 (1.1%) | 31 (2.1%) | 8 (1.3%) |
| Nausea | 10 (1.1%) | 28 (1.9%) | 6 (1.0%) |
| Myalgia | 3 (0.3%) | 26 (1.7%) | 4 (0.7%) |
| Injection site reactions (pain, erythema, bruising, hemorrhage, pruritus) | 11 (1.3%) | 23 (1.5%) | 12 (2.0%) |
| Influenza | 4 (0.5%) | 19 (1.3%) | 7 (1.1%) |
| Neutropenia | 4 (0.5%) | 15 (1.0%) | 5 (0.8%) |
| Tinea infections (tinea pedis, versicolor, cruris) | 2 (0.2%) | 15 (1.0%) | 3 (0.5%) |
Suicidal Ideation & Behaviour
During 12-wk randomised period: 1 subject in brodalumab group attempted suicide; 0 in placebo or ustekinumab. Through Week 52: 7/4,019 brodalumab (0.2 per 100 subject-years) vs 2/613 ustekinumab (0.4 per 100 subject-years). Overall programme: 34/4,464 (0.37 per 100 subject-years); 4 completed suicides.
Crohn’s Disease
One subject developed Crohn’s disease during psoriasis trials (excluded patients with active Crohn’s at baseline); led to discontinuation. Exacerbation of Crohn’s observed in other (non-psoriasis) trials. Contraindicated. Discontinue if Crohn’s develops.
Serious Infections
12-wk period: 0.5% brodalumab vs 0.2% placebo. One case of cryptococcal meningitis in brodalumab group. Overall through Week 52: exposure-adjusted rates similar between brodalumab and ustekinumab. Infections in 25.4% brodalumab vs 23.4% placebo (12-wk period).
Fungal Infections
Overall: 2.4% brodalumab vs 0.9% placebo. 12-wk period: 1.8% vs 0.9%. Primarily non-serious skin and mucosal Candida infections (oral 0.2%, genital 0.1%, esophageal 0.1% vs 0% placebo). Tinea infections: 1.0% vs 0.2%.
Neutropenia
12-wk period: 0.7% (any grade). Grade ≥3 (<1000/mm³): 0.5% brodalumab vs 0.2% ustekinumab vs 0% placebo. ANC reduction: 6.8% vs 3.3% vs 3.6%. Long-term: exposure-adjusted rate 0.4 per 100 subject-years (0.1 ≥Grade 3). No serious infections associated.
Long-term Safety (Through Wk 52 and Beyond)
Through Week 52: exposure-adjusted serious AE rates similar between brodalumab and ustekinumab. Through end of trial: exposure-adjusted rates similar to 52-week period. 3,755/4,464 subjects exposed ≥1 year.
Immunogenicity (52-week period): Approximately 3% of subjects treated with brodalumab developed antibodies to brodalumab through the 52-week treatment period. Of subjects who developed antibodies, none had antibodies classified as neutralising. However, the assay to test for neutralising antibodies had limitations detecting them in the presence of brodalumab; therefore, the incidence of neutralising antibody development could be underestimated. Source: PI Section 6.2, BLA 761032 (Ref ID 4056898).
| Population | Data / Recommendation |
|---|
| Pregnancy | No human data. Human IgG crosses the placental barrier; brodalumab may be transmitted to developing fetus. Animal data: no adverse developmental effects (embryofetal toxicity, malformations, morphological/functional/immunological development) in infants from cynomolgus monkeys administered brodalumab SC weekly at doses up to 26× MRHD (90 mg/kg/week) from organogenesis through parturition. Use only if benefit outweighs risk. |
| Lactation | Brodalumab detected in milk of lactating cynomolgus monkeys. No human data on presence in breast milk, effects on breastfed infant, or milk production. Consider developmental/health benefits of breastfeeding along with mother’s clinical need and potential adverse effects on infant. |
| Paediatric | Safety and effectiveness not evaluated in patients <18 years. Not approved for paediatric use. |
| Geriatric (≥65 years) | 192/3,066 (6%) subjects were ≥65 years; no subjects were ≥75 years. No differences in safety or efficacy vs younger subjects observed; however, numbers insufficient to determine whether elderly respond differently. Population PK: age does not significantly influence clearance of brodalumab. |
| Hepatic Impairment | No dedicated trials conducted. No dose adjustment recommended in label. |
| Renal Impairment | No dedicated trials conducted. No dose adjustment recommended in label. |
Carcinogenicity
Animal studies to evaluate carcinogenic potential of brodalumab have not been conducted. Published literature is mixed: some data suggest IL-17A promotes cancer cell invasion (potential beneficial effect of blockade); other data suggest IL-17A promotes T-cell mediated tumour rejection (potential adverse effect). Relevance for humans is unknown. Source: PI Section 13.1, BLA 761032.
Genotoxicity / Mutagenicity
Animal studies to evaluate mutagenic potential have not been conducted. Source: PI Section 13.1, BLA 761032.
Reproductive Toxicity / Fertility
Cynomolgus monkeys: no effects on fertility parameters (reproductive organs or sperm analysis) following SC administration up to 90 mg/kg/week for 6 months (26× MRHD). Monkeys were not mated to evaluate fertility effects directly. Embryofetal/postnatal study: no teratogenicity at 26× MRHD. Source: PI Section 13.1, BLA 761032.
Cardiac Safety (QTc)
Not reported in PI. Formal QTc studies not described. Source: PI Section 13.1, BLA 761032.
Tmax (single dose)
~3 days
Following single SC 210 mg dose in plaque psoriasis subjects
Cmax — Single Dose
13.4 ± 7.3 mcg/mL
Mean (±SD) serum Cmax after single 210 mg SC dose
AUC — Single Dose
111 ± 64 mcg·day/mL
Mean (±SD) AUC after single 210 mg SC dose
Cmax — Steady State (Q2W)
20.6 ± 14.6 mcg/mL
Mean (±SD) Cmax at steady state (210 mg Q2W)
AUCτ — Steady State (Q2W)
227 ± 167 mcg·day/mL
Mean (±SD) AUC over 2-week dosing interval at steady state
Steady State Achieved
Week 4
Following multiple SC doses of 210 mg Q2W
Bioavailability (%F)
~55%
Following SC administration
Volume of Distribution (Vz/F)
8.9 ± 9.4 L
Mean (±SD) apparent Vz/F after single 210 mg SC dose
Clearance (CL/F)
3.0 ± 3.5 L/day
Mean (±SD) apparent total CL/F after single 210 mg SC dose; nonlinear elimination
Absorption: Following a single subcutaneous dose of 210 mg in subjects with plaque psoriasis, brodalumab reached peak mean serum concentration (Cmax) of 13.4 ± 7.3 mcg/mL by approximately 3 days post-dose. Following multiple subcutaneous doses of 210 mg Q2W, steady-state was achieved by Week 4. Brodalumab exhibited non-linear pharmacokinetics with exposures that increased greater than dose-proportionally over a dose range from 140 mg (~0.67× recommended dose) to 350 mg (~1.67× recommended dose) following SC administration. No dedicated food-effect or formulation PK studies were conducted (administered as SC injection).
Following a single subcutaneous administration of brodalumab 210 mg, the mean (±SD) apparent volume of distribution (Vz/F) was 8.9 ± 9.4 L, consistent with predominantly vascular and limited extravascular distribution typical of IgG2 monoclonal antibodies. Protein binding and tissue distribution data were not characterised in the PI or clinical pharmacology review.
| Parameter | Details |
|---|
| Primary metabolic pathway | The metabolic pathway of brodalumab has not been characterised. As a human monoclonal IgG2 antibody, brodalumab is expected to be degraded into small peptides and amino acids via catabolic pathways in a manner similar to endogenous IgG. |
| Enzymes involved | Not applicable (proteolytic catabolism, not CYP-mediated) |
| Major metabolites | Small peptides and amino acids (non-active breakdown products) |
The mean (±SD) apparent total clearance (CL/F) was 3.0 ± 3.5 L/day after a single 210 mg SC dose. The clearance of brodalumab increased with decreasing doses due to nonlinear (target-mediated) elimination. The terminal half-life is not explicitly stated in the PI. The metabolic pathway has not been characterised; route of excretion (renal vs. fecal) is not reported. No dedicated renal or hepatic impairment PK trials were conducted.
| Population | PK Finding |
|---|
| Geriatric (≥65 years) | Population PK analysis indicated that age did not significantly influence the clearance of brodalumab. Subjects ≥65 years had similar clearance compared to subjects <65 years. |
| Body Weight | Brodalumab trough concentrations were lower in subjects with higher body weight. Weight effect is captured in population PK; fixed dosing without weight-based adjustment per approved label. |
| Hepatic Impairment | No dedicated trials conducted; not studied. |
| Renal Impairment | No dedicated trials conducted; not studied. |
| Paediatric | Not studied. |
| Race/Ethnicity | Not reported in PI as a significant covariate in population PK. |
| Interacting Drug/Class | Mechanism | Direction of Effect | Magnitude | Clinical Recommendation |
|---|
| CYP3A4 substrates (e.g., midazolam) | Brodalumab modulates serum cytokine levels (IL-17 pathway); cytokines (IL-1, IL-6, IL-10, TNFα, IFN) can alter CYP450 enzyme formation. Brodalumab treatment normalises inflammatory-state CYP450 suppression. | Increased substrate exposure (midazolam AUC increased) | Midazolam AUC increased by 24% one week following single 210 mg SC brodalumab dose | Upon initiation or discontinuation of brodalumab in patients receiving CYP450 substrates with narrow therapeutic index (e.g., warfarin, cyclosporine), consider monitoring for effect or drug concentration and consider dosage modification of the CYP450 substrate. |
| Live vaccines | Immunosuppression | Potential reduced vaccine immunogenicity; risk of vaccine-associated infection | Not quantified | Avoid use of live vaccines in patients treated with brodalumab. No data available on immune response to live or inactive vaccines during treatment. |
Brodalumab is a fully human monoclonal IgG2 antibody that selectively binds to human IL-17RA and inhibits its interactions with cytokines IL-17A, IL-17F, IL-17C, the IL-17A/F heterodimer, and IL-25. IL-17RA is a protein expressed on the cell surface and is a required component of receptor complexes utilised by multiple IL-17 family cytokines. Blocking IL-17RA inhibits IL-17 cytokine-induced responses including the release of pro-inflammatory cytokines and chemokines involved in psoriatic plaque formation. This receptor-level approach distinguishes brodalumab from secukinumab and ixekizumab, which target the individual IL-17A ligand rather than the shared receptor subunit.
Elevated levels of IL-17A, IL-17C, and IL-17F are found in psoriatic plaques. Serum IL-17A levels, measured at Weeks 12, 24, and 48 during treatment with brodalumab 210 mg Q2W, were higher than baseline levels in subjects with moderate-to-severe plaque psoriasis — consistent with receptor blockade causing accumulation of free cytokine in serum. The relationship between this pharmacodynamic activity and the mechanism(s) by which brodalumab exerts its clinical effects is unknown. Formal dose-response or exposure-response analyses are not reported in the PI.
Approved Dose
210 mg
Per injection; subcutaneous; single-dose prefilled syringe
Route
Subcutaneous
Thigh, abdomen (not around navel), or outer upper arm (if caregiver-administered)
Frequency
Q2W (after loading)
Weeks 0, 1, 2 (loading), then every 2 weeks
Duration
Reassess at 12–16 wks
Discontinue if adequate response not achieved by 12–16 weeks
Standard Dosing
- Loading doses: 210 mg SC at Weeks 0, 1, and 2
- Maintenance: 210 mg SC every 2 weeks (Q2W)
- If adequate response not achieved after 12–16 weeks, consider discontinuing therapy
- Continued treatment beyond 16 weeks in non-responders is not likely to result in greater success
Dose Modifications
- Renal impairment: No dedicated studies; no dose adjustment specified in label
- Hepatic impairment: No dedicated studies; no dose adjustment specified in label
- Paediatric: Not approved; no dosing established
- Elderly (≥65 years): No dose adjustment required (similar PK to younger subjects)
- Body weight: Fixed 210 mg dose regardless of weight (trough concentrations lower in higher-weight patients but no adjustment mandated)
Preparation & Administration
- Allow prefilled syringe to reach room temperature (~30 minutes) before injecting; do not warm by other means
- Visually inspect: clear to slightly opalescent, colourless to slightly yellow solution; few translucent to white amorphous particles may be present
- Do not use if cloudy, discoloured, or foreign matter present; do not use if dropped on hard surface
- Single-dose only; inject the full 1.5 mL (210 mg)
- Insert needle at 45–90 degrees; inject with slow constant pressure
- Rotate injection sites; do not inject into tender, bruised, red, hard, thick, scaly skin or psoriatic plaques
- Discard used syringe in FDA-cleared sharps container
Missed Dose
- No specific missed dose instructions provided in the prescribing information
- General guidance: administer as soon as possible, then resume on the regular schedule
- Instruct patients to read the Instructions for Use and consult their healthcare provider if unsure
CONTRAINDICATION — Crohn’s Disease: SILIQ is contraindicated in patients with Crohn’s disease because SILIQ may cause worsening of disease. Crohn’s disease occurred in one subject during psoriasis trials; exacerbation of Crohn’s disease was observed with SILIQ use in other trials. Discontinue SILIQ if the patient develops Crohn’s disease while taking SILIQ. Source: PI Section 4 and 5.5, BLA 761032 (Ref ID 4056898).
Mandatory SILIQ REMS Enrolment Before Prescribing: Prescribers must be certified with the SILIQ REMS program. Patients must sign a Patient-Prescriber Agreement Form. Pharmacies must be certified and may only dispense to authorised patients. Patients must carry the SILIQ Patient Wallet Card at all times. Information: www.SILIQREMS.com or 855-511-6135.
| Assessment / Requirement | Recommendation |
|---|
| TB Screening | Evaluate all patients for TB infection prior to initiating brodalumab. Do not administer to patients with active TB. Initiate treatment for latent TB before starting. Consider anti-TB therapy in patients with past latent/active TB if adequate treatment cannot be confirmed. Monitor for signs/symptoms of active TB during and after treatment. |
| Psychiatric Assessment / Depression | Review personal and family history of depression, suicidal ideation, and behaviour prior to prescribing. Weigh risks vs benefits in patients with history of depression or suicidality. Enrol patient in SILIQ REMS (patient-prescriber agreement required). |
| Crohn’s Disease Screening | Screen for pre-existing Crohn’s disease; contraindicated if present. Assess for signs/symptoms of IBD. |
| Live Vaccinations | Complete all required live vaccinations prior to initiating treatment. Avoid live vaccines during treatment. |
| Chronic/Recurrent Infections | Consider risks and benefits prior to prescribing in patients with chronic or recurrent infections. Instruct patients to seek medical advice if signs/symptoms of infection develop. |
| Drug/Class | Effect | Clinical Action |
|---|
| CYP450 substrates with narrow therapeutic index (e.g., warfarin, cyclosporine) | Brodalumab modulates cytokine levels which can alter CYP450 enzyme activity; midazolam (CYP3A4 substrate) AUC increased 24% following single brodalumab dose | Upon initiation or discontinuation of brodalumab, monitor therapeutic effect or drug concentration of co-administered narrow therapeutic index CYP450 substrates; consider dosage modification of the CYP450 substrate |
| Parameter | Recommendation |
|---|
| Injection sites | Thigh; abdomen (except 2-inch area around navel); outer upper arm (only if caregiver-administered) |
| Site rotation | Choose a different site each time; do not use same spot on an injection site used previously |
| Sites to avoid | Tender, bruised, red, hard, thick, scaly skin; areas with scars or stretch marks; active psoriatic plaques |
| Needle angle | Insert at 45 to 90 degrees; pinch skin to create firm surface; keep skin pinched while injecting |
| Patient self-injection | Permitted when deemed appropriate by healthcare professional and after proper training in SC injection technique using prefilled syringe |
| Storage after removal from refrigerator | May store at room temperature up to 25°C (77°F) for maximum 14 days; do not return to refrigerator; discard after 14 days at room temperature |
| Syringe handling | Do not shake; do not remove gray needle cap until ready to inject; do not reuse; dispose in FDA-cleared sharps container |
| Item | Details |
|---|
| Application Number | BLA 761032 (Orig-1/S-000) |
| Application Type | Biologics License Application (BLA) 351(a) |
| Applicant | Valeant Pharmaceuticals North America LLC, Bridgewater, NJ 08807 USA |
| Original Developer | Amgen Inc. (AMG 827); Kyowa Hakko Kirin Co., Ltd. (KHK4827); licensed to Valeant Pharmaceuticals |
| Submission Date | 16-NOV-2015 |
| PDUFA Goal Date | 16-NOV-2016 |
| Actual Approval Date | 15-FEB-2017 |
| Review Division | Division of Dermatology and Dental Products (DDDP), Office of Drug Evaluation III (ODE3), OND, CDER |
| Review Type | Standard Review |
| Breakthrough Therapy Designation | Not reported in PI or Medical Review as granted |
| Fast Track Designation | Not reported |
| Orphan Drug Designation | Not applicable |
| Advisory Committee | Not reported as convened for this application |
| Medical Review Ref ID | Ref ID 4056898 |
| PI Ref ID | Ref ID 4056898; Issued February 2017 |
2009–2011
Early Development — AMG 827 / KHK4827
Brodalumab (AMG 827, co-developed by Amgen and Kyowa Hakko Kirin) entered Phase 1/2 development across multiple inflammatory indications including psoriasis, psoriatic arthritis, rheumatoid arthritis, asthma, and Crohn’s disease. IND filing initiated development programme.
2012–2014
Phase 3 AMAGINE Programme Initiated
Three pivotal Phase 3 trials — AMAGINE-1 (NCT01708590), AMAGINE-2 (NCT01708577), AMAGINE-3 (NCT01708603) — commenced in plaque psoriasis. Total enrolment 4,373 subjects.
2014–2015
Suicidal Ideation and Behaviour Signal Identified — Early Trial Termination
During the Phase 3 programme, suicidal ideation and behaviour (SIB) emerged as a safety signal; 4 completed suicides were identified in the psoriasis Phase 3 cohort. The sponsor terminated all ongoing clinical trials for brodalumab early across all indications. AstraZeneca (which had co-developed the drug with Amgen) decided not to pursue approval. Valeant Pharmaceuticals acquired global rights and continued BLA preparation for psoriasis.
16-NOV-2015
BLA 761032 Submitted — Valeant Pharmaceuticals
Standard review designation. PDUFA goal date: 16-NOV-2016. Application included data from AMAGINE-1, -2, -3 pivotal trials plus open-label extension data. Manufacturing: Valeant Pharmaceuticals Luxembourg S.à.r.l., U.S. License No. 2053.
AUG–NOV 2016
FDA Clinical Review — Initial Complete Response Recommended
FDA clinical reviewer (Gary T Chiang MD, MPH) initially recommended a Complete Response citing the SIB signal and unresolved benefit-risk. Key concern: 4 completed suicides; trial termination led to incomplete long-term safety data for the SIB signal. The REMS programme and mandatory Boxed Warning were negotiated as mitigation strategies.
NOV 2016
Complete Response Letter Issued
FDA issued a Complete Response Letter (CRL). The primary basis was the suicidal ideation and behaviour safety concern. Negotiations around REMS programme, Boxed Warning language, contraindication for Crohn’s disease, and restriction of indication to patients who have failed other systemic therapies followed.
15-FEB-2017
FDA Approval — BLA 761032 — SILIQ (brodalumab)
Approved with Boxed Warning (suicidal ideation and behavior), mandatory SILIQ REMS Program (prescriber certification, patient-prescriber agreement, certified pharmacy dispensing), and contraindication for Crohn’s disease. Indication restricted to adults who are candidates for systemic therapy or phototherapy and have failed to respond or have lost response to other systemic therapies.
Suicidal Ideation and Behaviour (SIB) Signal
The dominant review issue for BLA 761032. 4 completed suicides and 34 cases of SIB (0.37 per 100 subject-years) were observed across the psoriasis Phase 3 programme. The primary clinical reviewer initially recommended a Complete Response. Resolution: Approval granted with Boxed Warning (strongest FDA warning), mandatory REMS, and restriction to patients who have failed other systemic therapies. Causal relationship not definitively established. Brodalumab is the only biologic in dermatology to carry both a Boxed Warning and REMS for suicidality.
Crohn’s Disease Signal — Contraindication
Crohn’s disease occurred in one psoriasis subject and exacerbation was observed in non-psoriasis trials (where IBD was not excluded). Resolution: Contraindication for patients with Crohn’s disease was mandated in the final labelling (PI Section 4). Screening required before initiation.
Early Trial Termination — Data Completeness
AstraZeneca’s decision to exit the programme and the sponsor’s early termination of all brodalumab trials resulted in incomplete long-term safety data, particularly for the SIB signal. This contributed to the initial CRL. Resolution: Available long-term data through Week 52 and open-label extensions were deemed sufficient to characterise short-term benefit-risk with appropriate risk mitigation (REMS, Boxed Warning, restricted indication).
Indication Restriction — “Failed Other Systemic Therapies”
To support a favourable benefit-risk profile for a drug with a Boxed Warning for SIB, the FDA restricted the indication to patients who have failed to respond or have lost response to other systemic therapies. This positions brodalumab as a later-line therapy rather than a first-line biologic, reserving its use for patients with unmet need where the efficacy benefit more clearly outweighs the psychiatric risk.
Postmarketing Requirements: As a condition of approval, the SILIQ REMS Program (PMR) was required. The REMS mandates prescriber certification, patient-prescriber agreement forms, certified pharmacy dispensing, and patient wallet cards. Specific additional PMR or PMC study designs are not explicitly detailed in the prescribing information (Ref ID 4056898). Source: PI Section 17 / BLA 761032 (Ref ID 4056898).
| PMR/PMC | Study Type | Objective | Key Design Elements |
|---|
| SILIQ REMS Program (PMR) | Risk Evaluation and Mitigation Strategy | Mitigate risk of suicidal ideation and behaviour associated with brodalumab; ensure prescribers and patients are informed | Prescriber certification; patient-prescriber agreement; certified pharmacy dispensing only; patient wallet card; ongoing monitoring |
| Domain | Note |
|---|
| Benefit-Risk Conclusion | FDA determined that the benefits of brodalumab (high PASI 75, PASI 100, and sPGA 0/1 response rates including superiority over ustekinumab for complete clearance) outweigh the risks (SIB signal, Crohn’s disease, infections) in the restricted population of adults with moderate-to-severe plaque psoriasis who have failed other systemic therapies, provided the REMS programme is in place. |
| Labelling Negotiations | Boxed Warning text negotiated to reflect that SIB has occurred but causal relationship not definitively established. Indication restricted to patients who have failed other systemic therapies as a benefit-risk measure. Contraindication for Crohn’s disease added. |
| Immunogenicity Labelling | ~3% ADA incidence through 52 weeks; no neutralising antibodies detected (assay limitations noted). Included in PI Section 6.2. |
| Unique Safety Feature | Only biologic approved in dermatology (as of approval date) with both a Boxed Warning for suicidality and a mandatory REMS programme. Comparator class (secukinumab, ixekizumab) does not carry these restrictions. |
| Source Documents | FDA PI BLA 761032 (Ref ID 4056898); FDA Medical Review BLA 761032 (Gary T Chiang MD, MPH). Reference ID: 4056898. |