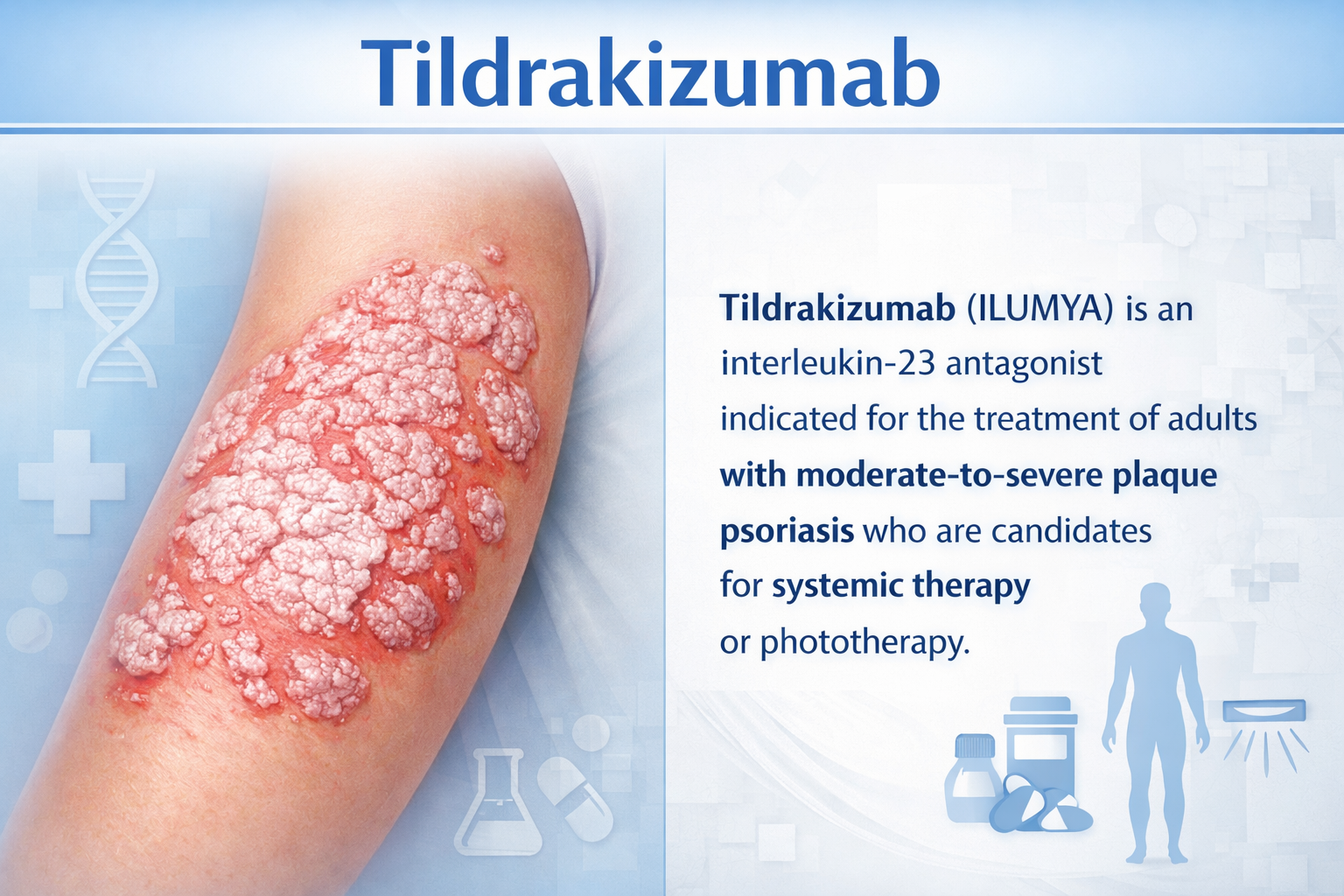
Indication
Moderate-to-Severe Plaque Psoriasis
Adults eligible for systemic therapy or phototherapy
Mechanism
IL-23p19 Antagonist
Humanized IgG1/κ monoclonal antibody · 147 kDa
Dose & Schedule
100 mg SC
Wk 0, Wk 4, then every 12 weeks (Q12W)
Phase 3 Programme
2 Pivotal Trials
P010 (N=772) + P011 (N=1,090) · 1,862 total
Mechanism of Action: Tildrakizumab binds selectively to the p19 subunit of IL-23 (Kd ~297 pM), preventing IL-23 from engaging its receptor (IL-23R). This blocks JAK2-mediated phosphorylation of STAT3, inhibiting homodimerisation and nuclear transcription of IL-17 and other pro-inflammatory cytokines central to psoriatic pathogenesis. Tildrakizumab does not bind IL-12 or the shared p40 subunit, distinguishing it from dual IL-12/23 inhibitors.
| Trial (ID) | NCT Number | Design | Arms & N Randomised | Duration | Primary Endpoint |
|---|
| Trial P003 (Phase 2) | Not reported | R, DB, 5-arm, dose-ranging, PC | Tildrakizumab 5/25/100/200 mg + placebo; N=270; ratio 1:2:2:2:1 | 72 weeks | PGA success at Wk 16; PASI 75 secondary |
| Trial P010 (Phase 3) | NCT01722331 | R, DB, PC, parallel-group, 3-part + LTE | Tildrakizumab 200 mg (N=308), 100 mg (N=309), Placebo (N=155) | 64 wk base + 192 wk extension | PASI 75 + PGA 0/1 at Wk 12 (co-primary) |
| Trial P011 (Phase 3) | NCT01729754 | R, DB, PC + active comparator (etanercept), 3-part + LTE | Tildrakizumab 200 mg (N=314), 100 mg (N=307), Etanercept 50 mg BIW (N=313), Placebo (N=156) | 52 wk base + 192 wk extension | PASI 75 + PGA 0/1 at Wk 12 (co-primary) |
| P05661 (Phase 1) | Not reported | Rising single-dose, IV, SAD in healthy volunteers | 0.1, 0.5, 3, 10 mg/kg IV + placebo; N=29 | Single dose | Safety, PK |
| P06306 (Phase 1) | Not reported | Non-randomized, open-label, race/ethnicity PK | 50, 200, 400 mg SC + 10 mg/kg IV; N=59 | Single dose | PK by race (Japanese, Chinese, White) |
| P009 (Phase 1) | Not reported | R, fixed-sequence, open-label, DDI (CYP450 cocktail) | 200 mg SC × 2 doses + probe cocktail; N=20 | 2 doses of tildrakizumab | CYP inhibition assessment; relative bioavailability PFS vs. autoinjector |
| Drug (INN · Brand) | Target | Formulation | US Approval | Approved Population |
|---|
| Tildrakizumab-asmn · ILUMYA | IL-23p19 | 100 mg SC Q12W | March 2018 | Adults, mod-severe plaque psoriasis |
| Ustekinumab · STELARA | IL-12/23 p40 | 45/90 mg SC Q12W | September 2009 | Adults & adolescents ≥12 yr |
| Secukinumab · COSENTYX | IL-17A | 300 mg SC Q4W (after loading) | January 2015 | Adults, mod-severe plaque psoriasis |
| Ixekizumab · TALTZ | IL-17A | 80 mg SC Q4W (after loading) | March 2016 | Adults, mod-severe plaque psoriasis |
| Guselkumab · TREMFYA | IL-23p19 | 100 mg SC Q8W (after loading) | July 2017 | Adults, mod-severe plaque psoriasis |
| Risankizumab · SKYRIZI | IL-23p19 | 150 mg SC Q12W | April 2019 | Adults, mod-severe plaque psoriasis |
| Parameter | Detail |
|---|
| Proprietary Name | ILUMYA |
| INN / Established Name | Tildrakizumab-asmn |
| Code Names (development) | SCH 900222; MK-3222 |
| Manufacturer / Applicant | Merck Sharp and Dohme Corp. (MSD) |
| Drug Product Manufacturer | MSD Ireland, Dublin Road, Carlow, Ireland |
| Pharmacologic Class | Interleukin-23 antagonist |
| Molecular Type | Recombinant humanized IgG1/κ monoclonal antibody; 4 polypeptide chains (2 heavy chains 446 aa each, 2 light chains 214 aa each); expressed in CHO cell line |
| Molecular Weight | ~147 kDa |
| Dosage Form & Strength | 100 mg/mL solution for injection in single-dose prefilled syringe (1 mL extractable volume); 29G staked needle; passive needle guard safety device |
| Storage | 2°C–8°C (36°F–46°F); protect from light; do not freeze; room temperature (≤25°C/77°F) up to 30 days; dating period: 36 months |
| Inactive Ingredients | L-histidine, L-histidine hydrochloride monohydrate, sucrose 7%, polysorbate 80 0.05%, Water for Injection; pH ~5.8 (b)(4) |
| Boxed Warning | None |
| PI Revision Date | March 2018 (original approval labeling) |
| Application Type | BLA 351(a), Public Health Service Act — original BLA (not a biosimilar) |
| US License Number | 0002 |
Pooled pivotal trials: Trial P010 enrolled and randomised 772 subjects at 112 investigational sites; Trial P011 enrolled and randomised 1,090 subjects at 123 investigational sites. Demographics were generally balanced across arms within each trial. Trial P010 included Japanese sites (higher Asian proportion: 23–27%); Trial P011 did not include Japanese sites (Asian: 2–4%). Prior biologic exposure was higher in P010 (23%) vs. P011 (12–13%). Full Analysis Set (FAS): all randomised subjects who received ≥1 dose.
| Parameter | Tildrakizumab 200 mg (N=308) | Tildrakizumab 100 mg (N=309) | Placebo (N=155) |
|---|
| Age mean (SD), yr | 47 (13) | 46 (13) | 48 (14) |
| Age median, yr | 48 | 46 | 48 |
| Age range, yr | 18–76 | 18–82 | 19–76 |
| Age <65 yr, n (%) | 279 (91%) | 281 (91%) | 136 (88%) |
| Age ≥65 yr, n (%) | 29 (9%) | 28 (9%) | 18 (12%) |
| Male, n (%) | 226 (73%) | 207 (67%) | 100 (65%) |
| White, n (%) | 209 (68%) | 217 (70%) | 101 (65%) |
| Asian, n (%) | 83 (27%) | 70 (23%) | 42 (27%) |
| Black, n (%) | 8 (3%) | 12 (4%) | 6 (4%) |
| Other, n (%) | 8 (3%) | 10 (3%) | 6 (4%) |
| Weight mean (SD), kg | 89 (24) | 89 (24) | 88 (26) |
| Weight median, kg | 86 | 86 | 85 |
| Weight ≤90 kg, n (%) | 182 (59%) | 183 (59%) | 93 (60%) |
| Weight >90 kg, n (%) | 126 (41%) | 126 (41%) | 62 (40%) |
| Prior biologic therapy, n (%) | 71 (23%) | 71 (23%) | 35 (23%) |
| U.S. sites, n (%) | 126 (41%) | 131 (42%) | 69 (45%) |
| Parameter | Tildrakizumab 200 mg (N=314) | Tildrakizumab 100 mg (N=307) | Etanercept 50 mg (N=313) | Placebo (N=156) |
|---|
| Age mean (SD), yr | 45 (14) | 45 (14) | 46 (14) | 46 (12) |
| Age median, yr | 45 | 44 | 48 | 46 |
| Age range, yr | 19–80 | 19–80 | 19–81 | 20–76 |
| Age <65 yr, n (%) | 289 (92%) | 280 (91%) | 282 (90%) | 142 (91%) |
| Age ≥65 yr, n (%) | 25 (8%) | 27 (9%) | 31 (10%) | 14 (9%) |
| Male, n (%) | 225 (72%) | 220 (72%) | 222 (71%) | 112 (72%) |
| White, n (%) | 284 (90%) | 279 (91%) | 289 (92%) | 144 (92%) |
| Asian, n (%) | 14 (4%) | 9 (3%) | 10 (3%) | 3 (2%) |
| Black, n (%) | 8 (3%) | 7 (2%) | 8 (3%) | 1 (1%) |
| Other, n (%) | 8 (3%) | 12 (4%) | 6 (2%) | 8 (5%) |
| Weight mean (SD), kg | 88 (21) | 89 (22) | 88 (21) | 89 (23) |
| Weight median, kg | 86 | 88 | 86 | 86 |
| Weight ≤90 kg, n (%) | 180 (57%) | 176 (57%) | 180 (58%) | 90 (58%) |
| Weight >90 kg, n (%) | 134 (43%) | 131 (43%) | 133 (42%) | 66 (42%) |
| Prior biologic therapy, n (%) | 38 (12%) | 39 (13%) | 37 (12%) | 20 (13%) |
| U.S. sites, n (%) | 86 (27%) | 85 (28%) | 87 (28%) | 44 (28%) |
| Parameter | P010 · Tildra 100 mg (N=309) | P010 · Placebo (N=155) | P011 · Tildra 100 mg (N=307) | P011 · Etanercept (N=313) | P011 · Placebo (N=156) |
|---|
| PGA Minimal (1), n (%) | 0 | 0 | 2 (1%) | 0 | 0 |
| PGA Mild (2), n (%) | 1 (<1%) | 0 | 9 (3%) | 7 (2%) | 7 (5%) |
| PGA Moderate (3), n (%) | 206 (67%) | 111 (72%) | 196 (64%) | 193 (62%) | 91 (59%) |
| PGA Marked (4), n (%) | 95 (31%) | 41 (27%) | 95 (31%) | 103 (33%) | 52 (34%) |
| PGA Severe (5), n (%) | 7 (2%) | 2 (1%) | 5 (2%) | 7 (2%) | 5 (3%) |
| PASI mean (SD) | 20.0 (7.9) | 19.3 (7.1) | 20.5 (7.6) | 20.2 (7.4) | 20.0 (7.6) |
| PASI median | 17.2 | 17.2 | 18.4 | 18.4 | 18.3 |
| PASI range | 9–59.4 | 12–51.8 | 8.4–54.8 | 8.4–52.5 | 12–55.8 |
| BSA % mean (SD) | 30 (17) | 30 (17) | 34 (18) | 32 (17) | 31 (15) |
| BSA % median | 24 | 25 | 30 | 28 | 29 |
| Parameter | Tildrakizumab 100 mg Safety Pool (N=1,083) |
|---|
| Prior biologic therapy | 17.5% (82.5% biologic-naïve) |
| Prior conventional systemic therapy | 40.2% |
| Prior phototherapy | 31.6% |
| History of psoriatic arthropathy | 10.8% |
| Hypertension | 27.1% |
| Obesity | 6.8% |
| Hyperlipidemia | 6.4% |
| Hypercholesterolemia | 6.0% |
| Type 2 diabetes mellitus | 5.1% |
| Back pain history | 4.8% |
| Prior SCC/BCC skin cancer | <1% |
| Race: White | 81.8% |
| Male sex | 71.4% |
| Median age, yr (range) | 46 (18–82) |
| Age ≥65 yr | 8.9% |
| Mean baseline PASI (SD) | 20.0 (7.7) |
| Baseline PGA 3 (Moderate) | 65.9% |
| Baseline PGA 4 (Marked) | 33.7% |
| Median BSA involved | 26% (range 5–100%) |
Co-primary endpoints (Week 12): PASI 75 response rate AND PGA 0/1 response (with ≥2-grade improvement from baseline). Both doses statistically superior to placebo in both trials (p < 0.001 for all 4 comparisons). Missing data: Non-Responder Imputation (NRI) — primary method. Multiplicity controlled via sequential gatekeeping: PASI 75 → PGA 0/1 → PASI 90 → PASI 100. CMH test stratified by baseline weight (≤90 kg / >90 kg) and prior biologic therapy use (Yes/No).
PASI 75 — tildrakizumab 100 mg
PASI 75 — tildrakizumab 200 mg
PGA 0/1 — tildrakizumab 100 mg
PGA 0/1 — tildrakizumab 200 mg
p < 0.001 for both tildrakizumab doses vs. placebo on both co-primary endpoints (CMH test).
PASI 75 — tildrakizumab 100 mg
PASI 75 — tildrakizumab 200 mg
PASI 75 — etanercept 50 mg
PGA 0/1 — tildrakizumab 100 mg
PGA 0/1 — tildrakizumab 200 mg
PGA 0/1 — etanercept 50 mg
p < 0.001 for both tildrakizumab doses vs. placebo. Etanercept vs. placebo comparison not a primary regulatory endpoint in this context; no approved comparative efficacy claim. Note: Trial P012 (etanercept comparator) was dropped; no comparative efficacy claim against etanercept was pursued. Source: FDA Medical Review BLA 761067 (Ref ID 4233639), Table 25.
PASI 90 — Tildra 100 mg (P010)
PASI 90 — Tildra 100 mg (P011)
PASI 100 — Tildra 100 mg (P010)
PASI 100 — Placebo (P010)
PASI 100 — Tildra 100 mg (P011)
PASI 100 — Placebo (P011)
Source: FDA Medical Review BLA 761067 (Ref ID 4233639), Table 25 (Week 12 values). Wk 0 and Wk 4 values not explicitly tabulated in text; shown as null. Wk 8 values not explicitly tabulated; shown as null. Only Wk 12 values were explicitly stated in the text tables extracted. spanGaps: false applied.
Source: FDA Medical Review BLA 761067 (Ref ID 4233639), Table 25 (Week 12 values). P011 includes etanercept active comparator arm. Intermediate timepoints (Wk 4, Wk 8) not explicitly tabulated; shown as null.
Re-randomisation design (Part 3): PASI 75 responders at Week 28 were re-randomised 1:1 to continue tildrakizumab 100 mg Q12W vs. withdraw to placebo. Non-responders (PASI <50% at Week 28) were discontinued. Partial responders (PASI 50–<75%) were re-randomised to 100 or 200 mg Q12W.
PASI 75 at Wk 64 — continue tildrakizumab 100 mg
PASI 75 at Wk 64 — withdrawal to placebo
PGA 0/1 at Wk 64 — continue tildrakizumab 100 mg
PGA 0/1 at Wk 64 — withdrawal to placebo
Rebound: Defined as worsening beyond baseline PASI (>125% of baseline PASI) or new pustular/erythrodermic/inflammatory psoriasis within 2 months of stopping therapy. No rebound events were observed in either withdrawal group (100 mg → placebo; 200 mg → placebo) during Weeks 32–64 of Trial P010.
| Parameter | Specification |
|---|
| Analysis population | Full Analysis Set (FAS): all randomised subjects receiving ≥1 dose; subjects with missing baseline PGA/PASI imputed as non-responders |
| Primary missing data method | Non-responder imputation (NRI) |
| Sensitivity analyses | (1) LOCF; (2) Multiple imputation (MI); (3) Worst-case scenario (tildrakizumab missing = non-responder; placebo missing = responder) — tildrakizumab remained superior to placebo in all scenarios |
| Inference test | Cochran–Mantel–Haenszel (CMH) test stratified by baseline weight (≤90 vs. >90 kg) and prior biologic use (Yes/No) |
| Multiplicity control | Sequential gatekeeping: PASI 75 (200 mg) → PASI 75 (100 mg) → PGA 0/1 (200 mg) → PGA 0/1 (100 mg) → PASI 90 → PASI 100; FWER controlled at α = 0.05 |
| Per-Protocol (PP) population | Results consistent with FAS; PP PASI 75: P010 100 mg = 64%, P011 100 mg = 61% |
No Boxed Warning. ILUMYA does not carry a Boxed Warning in the approved prescribing information.
Safety Population (Controlled)
1,083
Subjects receiving tildrakizumab 100 mg in Phase 2/3 trials
Long-Term Exposure
469
Subjects with ≥24 months exposure; 672 with ≥12 months
Mean Treatment Duration (Base Period)
53.9 weeks
Updated mean 108.1 wk (base + extension); total 4,130 subj-yr
Most Common ADR
URI (14%)
Upper respiratory infections (nasopharyngitis, URTI, pharyngitis)
Serious Hypersensitivity (§5.1 PI): Cases of angioedema and urticaria reported in post-marketing and clinical trials. Contraindicated with prior serious hypersensitivity to tildrakizumab or excipients. Discontinue immediately and initiate appropriate therapy if serious reaction occurs.
Infections (§5.2 PI): May increase susceptibility to infections. Infections reported in 23% of tildrakizumab vs. ~22% of placebo subjects. Serious infection rate: 1.0 per 100 subj-yr (tildrakizumab 100 mg) vs. 0.91 (placebo). Do not initiate in patients with active clinically important infections until resolved or adequately treated. Exercise caution in patients with chronic/recurrent infection.
Tuberculosis (§5.3 PI): Screen all patients for latent and active TB before initiating. One confirmed TB case (vertebral TB, tildrakizumab 200 mg group) was drug-related. Among 55 subjects with latent TB receiving concurrent prophylaxis plus tildrakizumab, no active TB cases occurred over mean 56.5 weeks. Do not administer to patients with active TB. Consider anti-TB therapy prior to initiating in patients with prior untreated latent TB.
Immunizations (§5.4 PI): Complete all age-appropriate immunisations according to current guidelines before initiating. Avoid live vaccines during treatment. No data available on secondary transmission of infection from live vaccine or on efficacy/safety of live or inactivated vaccines in patients receiving tildrakizumab.
| Adverse Reaction | Tildrakizumab 100 mg (N=705) n (%) | Placebo (N=355) n (%) |
|---|
| Upper respiratory infections* | 98 (13.9%) | 41 (11.5%) |
| Injection site reactions† | 24 (3.4%) | 8 (2.2%) |
| Diarrhea | 13 (1.8%) | 5 (1.4%) |
| Fatigue‡ | 17 (2.4%) | 6 (1.7%) |
| Back pain‡ | 9 (1.3%) | 4 (1.1%) |
| Abdominal pain‡ | 9 (1.3%) | 2 (0.6%) |
| System Organ Class | Tildrakizumab 100 mg (N=1,083)
n (per 100 subj-yr) | Tildrakizumab 200 mg (N=1,041)
n (per 100 subj-yr) | Placebo (N=588)
n (per 100 subj-yr) | Etanercept 50 mg (N=313)
n (per 100 subj-yr) |
|---|
| Any SAE | 58 (5.81) | 67 (7.21) | 14 (6.4) | 20 (13.4) |
| Infections & infestations | 10 (1.00) | 15 (1.61) | 2 (0.91) | 2 (1.30) |
| Neoplasms* | 15 (1.50) | 10 (1.08) | 3 (1.37) | 5 (3.26) |
| Cardiac disorders | 6 (0.60) | 8 (0.86) | 1 (0.46) | 2 (1.30) |
| Gastrointestinal disorders | 8 (0.80) | 4 (0.43) | 1 (0.46) | 0 (0.0) |
| Musculoskeletal disorders | 3 (0.30) | 8 (0.86) | 1 (0.46) | 1 (0.65) |
| Hepatobiliary disorders | 3 (0.30) | 1 (0.11) | 0 (0.0) | 1 (0.65) |
| Nervous system disorders | 2 (0.20) | 5 (0.54) | 2 (0.91) | 2 (1.30) |
Seven total deaths occurred across all treatment groups during the base periods of pooled Phase 2/3 trials (P003, P010, P011). Five deaths in the tildrakizumab 100 mg group (causes: unknown, alcoholic cardiomyopathy and steatohepatitis — 2 events for these combined); 1 death in the tildrakizumab 200 mg group (aneurysm); 1 death in the etanercept/tildrakizumab 200 mg group (sepsis). None of the deaths was assessed as causally related to study product by the investigators. No deaths in the Phase 1 programme (6 trials).
| Period / Group | Discontinuation Rate | Notes |
|---|
| Tildrakizumab 100 mg (placebo-controlled period, Wk 0–12) | 0.6% | Lowest across all groups in this period |
| Tildrakizumab 200 mg (placebo-controlled period) | 1.3% | Cardiac disorders SOC most common (2/708: AF, CAD) |
| Placebo (placebo-controlled period) | 1.1% | — |
| Etanercept (placebo-controlled period) | 1.0% | — |
| Tildrakizumab 100 mg (base period exposure-adjusted) | 2.20 per 100 subj-yr | Neoplasms SOC: highest discontinuations (6 subjects) |
| Tildrakizumab 200 mg (base period exposure-adjusted) | 2.15 per 100 subj-yr | — |
| Placebo (base period exposure-adjusted) | 2.28 per 100 subj-yr | — |
| Etanercept (base period exposure-adjusted) | 5.87 per 100 subj-yr | Notably higher than tildrakizumab groups |
Anti-Drug Antibody (ADA) Incidence
6.5% of subjects developed anti-tildrakizumab antibodies by up to Week 64 of the base and extension periods. ADA incidence was measured using a validated immunoassay.
Neutralising Antibodies (nAb)
~40% of ADA-positive subjects (~2.5% of total population) had neutralising antibodies. Development of neutralising antibodies was associated with lower serum tildrakizumab concentrations and reduced clinical efficacy (exposure–response impact confirmed in PK analysis).
Carcinogenicity
No carcinogenicity or mutagenicity studies conducted. Applicant provided a risk assessment using published literature. Nonclinical information does not suggest increased carcinogenic risk. Relevance of animal models to humans is unknown.
Genotoxicity
No genetic toxicology studies conducted. As a monoclonal antibody targeting a soluble cytokine, direct genotoxic potential is not anticipated by mechanism.
Reproductive & Developmental Toxicology (NHP)
Embryofetal study (cynomolgus monkeys, GD 20–118): NOAEL 300 mg/kg (79× MRHD by AUC). Pre/postnatal study: 4/14 high-dose infants died within 15 days; 2 deaths possibly tildrakizumab-related (viral infection pattern on histopathology). NOAEL maternal: 100 mg/kg (30× MRHD); NOAEL developmental: 10 mg/kg (3× MRHD). Tildrakizumab crosses placenta in monkeys.
Repeat-Dose Toxicology (NHP)
3-month study (SC 0/40/140 mg/kg; IV 140 mg/kg Q2W): No significant adverse effects; NOAEL = 140 mg/kg. 9-month study (SC 0/10/100 mg/kg Q2W): No significant adverse effects; NOAEL = 100 mg/kg (45× MRHD by AUC). Cynomolgus monkey was the only pharmacologically relevant species.
Bioavailability (%F)
73–80%
Absolute SC bioavailability
Tmax (median)
~6 days
After single SC 100 mg dose
Steady-State Cmax
8.1 mcg/mL
Geometric mean (CV% 34%) at Week 16
Steady-State Ctrough (Wk 16)
1.22–1.47 mcg/mL
Mean ±SD range across arms
Volume of Distribution (Vd)
10.8 L
Geometric mean (CV% 24%)
Systemic Clearance (CL)
0.32 L/day
Geometric mean (CV% 38%)
Half-life (t½)
~23 days
Geometric mean (CV% 23%)
Steady State Achieved
Week 16
Wk 0 → Wk 4 → Q12W schedule
Dose Proportionality
Linear
50–200 mg SC (0.5× to 2× approved dose)
SC absorption: Absolute bioavailability 73–80% following SC injection. Median Tmax ~6 days. Dose proportionality demonstrated across 50–200 mg. No formal food-effect study conducted (SC route, not applicable). Relative bioavailability: PFS and PFS-housed autoinjector were shown to be bioequivalent in Phase 1 trial P009.
Volume of distribution 10.8 L (geometric mean, CV% 24%), consistent with predominantly intravascular/tissue distribution typical of a large IgG1 monoclonal antibody. Specific tissue distribution not studied in humans. Tildrakizumab crosses the placenta in cynomolgus monkeys (infant/maternal serum ratios documented); breast milk excretion was minimal (0.09–0.2% of serum concentration in monkeys).
| Parameter | Detail |
|---|
| Metabolic pathway | Proteolytic catabolism to small peptides and amino acids via endogenous IgG catabolic pathways; no hepatic CYP450-mediated metabolism expected |
| Primary elimination route | Catabolism (monoclonal antibody); renal filtration not applicable at this MW (~147 kDa) |
| CYP involvement | None anticipated (biologic, not substrate for CYP enzymes) |
| Dedicated mass balance study | Not conducted |
| Population/Factor | Effect on PK | Clinical Action |
|---|
| Body weight (higher) | Higher weight associated with lower tildrakizumab concentrations (population PK analysis) | No dose adjustment required; effect not clinically significant at approved dose |
| Age (≥18 yr) | No clinically meaningful differences across age range studied | No dose adjustment |
| Race/Ethnicity | Phase 1 study P06306: Japanese, Chinese, White subjects — no clinically significant differences in PK | No dose adjustment |
| Renal impairment | Dedicated study not conducted; mAbs not renally cleared | No dose adjustment anticipated |
| Hepatic impairment | Dedicated study not conducted; hepatic CYP metabolism not applicable | No dose adjustment anticipated |
| Sex | Not reported as a significant covariate in population PK | No dose adjustment |
DDI rationale: IL-23 inhibition may normalise IL-6-driven CYP450 suppression in psoriasis, potentially restoring CYP450 activity. Evaluated at tildrakizumab 200 mg SC (twice the approved dose) after 2 doses at Weeks 0 and 4.
| Probe Substrate | CYP Isoform | Effect (Tildrakizumab 200 mg) | Magnitude | Clinical Recommendation |
|---|
| Midazolam | CYP3A4 | No clinically significant change | Not reported as significant | No action required |
| Dextromethorphan | CYP2D6 | Modest increase in AUC | ~20% increase in AUCinf | Unlikely to require dose adjustment for CYP2D6 substrates |
| Caffeine | CYP1A2 | No clinically significant change | Not reported as significant | No action required |
| Warfarin | CYP2C9 | No clinically significant change | Not reported as significant | No action required |
| Omeprazole | CYP2C19 | No clinically significant change | Not reported as significant | No action required |
Live vaccines: As an immunomodulatory biologic, concomitant use of live vaccines is contraindicated. No formal DDI study with vaccines; based on pharmacological class effect.
Tildrakizumab is a humanized recombinant IgG1/κ monoclonal antibody that selectively binds the p19 subunit of human IL-23 with a dissociation constant (Kd) of approximately 297 pM. The epitope is primarily on the p19 subunit with limited interaction with the p40 subunit, preserving IL-12 activity. Binding prevents IL-23 from engaging its receptor (IL-23R), blocking JAK2-mediated phosphorylation of STAT3. This abrogates STAT3 homodimerisation, nuclear translocation, and transcription of pro-inflammatory cytokines including IL-17A, IL-17F, and IL-22, which drive keratinocyte hyperproliferation, epidermal thickening, and dermal inflammation in psoriasis. Tildrakizumab does not bind human IL-12 (confirmed by surface plasmon resonance and indirect ELISA), nor does it bind murine or rat IL-23.
In vitro neutralisation of IL-23 biological activity demonstrated IC₅₀ values of 59–187 pM across three orthogonal cell-based assay platforms (BAF/3 transfected cells, primary human splenocytes, KIT225 T-cell line). Tildrakizumab binds cynomolgus monkey IL-23 with higher affinity (Kd ~47 pM). Exposure–response analyses (population PK/PD) demonstrated a positive correlation between tildrakizumab trough concentrations and efficacy (PASI 75 and PGA 0/1 response rates) at Week 12, supporting the selected dosing regimen. Subjects with neutralising anti-drug antibodies had lower serum tildrakizumab concentrations and reduced clinical response, consistent with PK-mediated immunogenicity impact.
Approved Dose
100 mg SC
Per single-dose prefilled syringe
Route
Subcutaneous
HCP-administered (no self-injection approved at launch)
Frequency
Wk 0, 4, Q12W
Initial loading at Wk 0 and Wk 4; maintenance Q12W
Duration
Continuous
No defined treatment limit; reassess periodically
Standard Dosing
- 100 mg SC at Week 0 (initial dose)
- 100 mg SC at Week 4 (loading dose)
- 100 mg SC every 12 weeks (Q12W) thereafter
- No approved dose above 100 mg for plaque psoriasis
- 200 mg dose not approved (higher AE burden without superior efficacy at label)
Dose Modifications
- Renal impairment: no dose adjustment (no dedicated PK study; mAb catabolism)
- Hepatic impairment: no dose adjustment (no dedicated PK study)
- Elderly (≥65 yr): no dose adjustment
- Body weight: no weight-based dosing (flat dosing regimen)
- No dose reduction criteria defined in PI
Preparation & Administration
- Remove from refrigerator; allow 30 min to reach room temperature
- Inspect visually: clear to slightly opalescent, colourless to slightly yellow; discard if discoloured, cloudy, or contains particles
- Administer SC only: abdomen, thighs, or upper arm
- Avoid 2 inches around navel; avoid tender/bruised/erythematous/indurated skin
- Avoid psoriasis-affected skin, scars, stretch marks
- Rotate injection sites; do not inject into same site twice in a row
- Do not shake the syringe
Missed Dose
- Administer as soon as possible if a dose is missed
- Resume Q12W schedule from the date of the missed dose administration
- Do not double-dose
- No specific bridging required unless extended interruption due to serious infection
| Condition | Specification |
|---|
| Refrigerated storage | 2°C–8°C (36°F–46°F); keep in original carton; protect from light |
| Room temperature | Stable up to 30 days at ≤25°C (77°F); once removed from refrigerator, do not return |
| Dating period (drug product) | 36 months at 2–8°C |
| Do not | Freeze; shake; expose to light without carton |
Contraindication (PI §4): Prior serious hypersensitivity reaction to tildrakizumab or to any of the excipients of ILUMYA. Excipients: L-histidine, L-histidine hydrochloride monohydrate, sucrose 7%, polysorbate 80 0.05%, Water for Injection.
TB screening required before first dose: Evaluate all patients for latent and active TB. Initiate treatment of latent TB prior to initiating tildrakizumab.
Immunisations before initiating: Complete all age-appropriate immunisations per current guidelines before first dose. Avoid live vaccines during treatment.
Active infection: Do not initiate tildrakizumab in patients with any clinically important active infection until resolved or adequately treated.
| Interacting Drug/Class | Mechanism | Recommendation |
|---|
| Live vaccines | Immunosuppression — potential vaccine failure or disease transmission | Contraindicated; avoid during treatment |
| CYP2D6 substrates (narrow TI) | Modest (~20%) CYP2D6 AUC increase with 200 mg; effect at 100 mg unknown but likely smaller | Clinical significance uncertain; no dose adjustment requirement identified |
| CYP3A4, 1A2, 2C9, 2C19 substrates | No clinically significant interaction detected at 200 mg | No dose adjustment required |
| Parameter | Recommendation |
|---|
| Injection technique | Pinch skin and insert needle at 45–90° angle; inject full 1 mL volume; apply pressure after withdrawal |
| Needle guard activation | Passive safety device activates automatically on needle withdrawal; do not recap |
| Site rotation | Rotate across abdomen, thigh, and upper arm; document prior sites |
| Disposal | Dispose of used prefilled syringe immediately in an appropriate sharps container |
| Caregiver training | HCP should train patient/caregiver on SC injection technique before any self-administration |
| Parameter | Detail |
|---|
| Application Number | BLA 761067 |
| Application Type | BLA 351(a), Public Health Service Act — Original BLA (not a biosimilar or supplement) |
| Applicant | Merck Sharp and Dohme Corp. |
| Original Developer | Schering-Plough (acquired by Merck 2009); developed as SCH 900222 / MK-3222 |
| Submission Date | March 23, 2017 |
| PDUFA Goal Date | March 23, 2018 |
| Actual Approval Date | March 2018 (review completed March 13, 2018) |
| Review Division | Division of Dermatology and Dental Products (DDDP), Office of Drug Evaluation III (ODE III) |
| Review Type | Standard Review |
| Breakthrough Therapy Designation | Not designated |
| Fast Track Designation | Not designated |
| Orphan Drug Designation | Not applicable |
| Advisory Committee | Not convened (no advisory committee meeting held for this application) |
| Medical Review Ref ID | 4233639 (Multi-Disciplinary Review and Evaluation) |
| Statistical Review Ref ID | 4237390 |
| CDTL Review Ref ID | 4236563 |
| Review Team | Clinical: Melinda McCord MD, Kevin Clark MD; CDTL: Gordana Diglisic MD; Statistics: Matthew Guerra PhD, Mohamed Alosh PhD; Project Manager: Dawn Williams |
APRIL 1, 2008
Pre-IND Meeting
Initial FDA meeting prior to IND filing to discuss development programme for SCH 900222 / MK-3222.
SEPTEMBER 8, 2008
IND 101389 Opened
IND opened for tildrakizumab (code names SCH 900222 and MK-3222) under development by Schering-Plough/Merck.
JUNE 1, 2011
Guidance Meeting
FDA advised applicant on need to characterise PK of to-be-marketed prefilled syringe formulation and on optimal PK/immunogenicity sampling timepoints for Phase 3 trials. Human factors study for PFS recommended.
APRIL 11, 2012
End-of-Phase 2 (EOP2) Meeting
Applicant and Agency agreed to co-primary endpoints: PASI 75 and PGA 0/1 (≥2-grade improvement) at Week 12. Agency provided guidance on multiplicity control, randomisation stratification, missing data handling (NRI primary), and MACE adjudication. Three Phase 3 protocols (P010, P011, P012) proposed.
JUNE 8 – JULY 18, 2012
Special Protocol Assessments (SPA)
SPAs submitted for Trials P010, P011, and P012. Agreement letters issued July 18, 2012. Agency reiterated comments on multiplicity, placebo arm continuity to Week 28 in P011/P012, and ITT population use.
NOVEMBER 24, 2015
Amended Protocols — PASI 90/100 Added; P012 Dropped
Applicant amended P010/P011 protocols to add PASI 90 and PASI 100 as key secondary endpoints with sequential gatekeeping. Applicant confirmed abandonment of Trial P012 and withdrawal of comparative efficacy claim versus etanercept. FDA concurred with amendments in advice letter December 29, 2015.
DECEMBER 2015 – MAY 2016
Agreed Initial Paediatric Study Plan (iPSP)
iPSP submitted December 11, 2015; agreed iPSP May 20, 2016. Partial waiver granted for <6 yr (low prevalence); deferral granted for 6–18 yr until adult approval.
MARCH 23, 2017
BLA 761067 Submitted
Original BLA submitted in eCTD format by Merck Sharp & Dohme Corp. Standard review designation. Two pivotal Phase 3 trials (P010, P011) plus Phase 2 data (P003) and 6 Phase 1 trials submitted.
OCTOBER 2, 2017
120-Day Safety Update Report (SUR) Received
Updated mean treatment duration 108.1 weeks; total exposure 4,130.24 subject-years (base + extension).
MARCH 13, 2018
Multi-Disciplinary Review Signed (Ref ID 4233639)
All reviewers recommended approval. CDTL (Gordana Diglisic MD) and clinical team (Melinda McCord MD, Kevin Clark MD) concluded data support approval. Statistical reviewer (Matthew Guerra PhD) confirmed statistical analysis of efficacy supports approval.
MARCH 2018
FDA Approval — BLA 761067
Approved indication: ILUMYA (tildrakizumab-asmn) injection 100 mg/mL for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. U.S. License No. 0002. Drug Product manufactured at MSD Ireland, Dublin Road, Carlow, Ireland. Dating period: 36 months at 2–8°C.
Statistical Multiplicity — Phase 3 Protocol
Agency initially identified that the applicant’s proposed stepwise multiplicity control method did not fully control the Type I error rate (noted at EOP2). Resolved by adoption of a sequential gatekeeping approach covering both doses and both co-primary endpoints (PASI 75 → PGA 0/1 → PASI 90 → PASI 100), agreed via SPA process and confirmed in December 2015 advice letter.
Missing Data Handling
Agency required justification for LOCF as a sensitivity analysis (cited limited evidence base for LOCF). Resolved: applicant retained NRI as primary method and added multiple imputation (MI) as second sensitivity; FDA statistical reviewer added worst-case scenario analysis which confirmed superiority under all imputation frameworks.
Trial P012 Discontinuation
Agency recommended placebo arm in P012 through Week 28 to avoid interpretation difficulties. Applicant cancelled P012 and withdrew comparative efficacy claim against etanercept entirely. FDA accepted this in April 2015 guidance meeting; Trial P011 data still included etanercept as active comparator through Wk 28 without a regulatory comparative claim.
TB Case & Safety Signal
One drug-related vertebral TB case (tildrakizumab 200 mg) required labelled precaution. The TB screening requirement and monitoring language were added to PI §5.3. The 100 mg approved dose had no TB cases among the 55 latent TB subjects receiving concurrent prophylaxis during trials.
Neoplasm Signal — Ongoing Monitoring
Malignancies (including melanoma, bladder cancer, breast cancer, haematologic) assessed as possibly related to treatment necessitated inclusion of immunogenicity risk context in labelling and post-marketing surveillance plan. No increase in total malignancy rate relative to background psoriasis population risk was concluded.
Paediatric Assessment
Agency required agreed iPSP per PREA requirements. Partial waiver granted for <6 yr (extremely low prevalence of severe psoriasis); paediatric studies deferred for 6–18 yr pending adult approval. Studies remain outstanding as post-marketing paediatric commitments.
PMC summary: No CMC-specific postmarketing commitments (PMCs) from the CMC review team. Stability protocols approved for annual assessment and for extending expiration dating (21 CFR 601.12). Product is exempt from lot release per 21 CFR 601.2(a). Paediatric study requirements outstanding per iPSP agreement.
| PMR/PMC | Study Type | Objective | Key Design Elements |
|---|
| Paediatric deferral (iPSP) | Efficacy & safety trial(s) | Evaluate tildrakizumab in paediatric plaque psoriasis ages 6–<18 yr | Deferred until adult approval confirmed; design TBD; partial waiver for <6 yr |
| Annual stability protocols | Drug Substance & Drug Product stability | Extend expiration dating; trend release results | Annual stability per approved protocol; 21 CFR 601.12 compliance |
| None (CMC) | — | No additional CMC PMCs identified by CMC review team | N/A |
| Domain | Note |
|---|
| Benefit-risk conclusion | Available safety and efficacy data support approval. Both pivotal trials met all co-primary and key secondary endpoints. Safety profile acceptable for target population with appropriate labelling of TB, infection, and hypersensitivity risks. |
| Approved dose selection | 100 mg approved; 200 mg dose not approved — no meaningful efficacy advantage over 100 mg with higher SAE burden (particularly musculoskeletal and infection SAEs) at 200 mg. |
| Immunogenicity labelling | ADA incidence (6.5%) and nAb impact (reduced PK and efficacy) required labelling per FDA biologics guidance. Included in PI §6.1. |
| REMS | No Risk Evaluation and Mitigation Strategy (REMS) required. No safety issues identified that could not be managed through routine labelling. |
| Advisory Committee | None convened. Application reviewed entirely through normal DDDP/ODE III clinical review process. |
| Lot release exemption | ILUMYA exempted from lot release per 601.2(a) as a specified product. U.S. License No. 0002. |
| Source document Ref IDs | Multi-Disciplinary Review: Ref ID 4233639 | Statistical Review Memo: Ref ID 4237390 | CDTL Memo: Ref ID 4236563 |